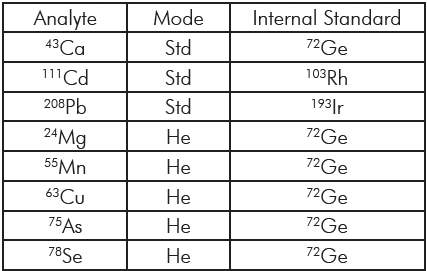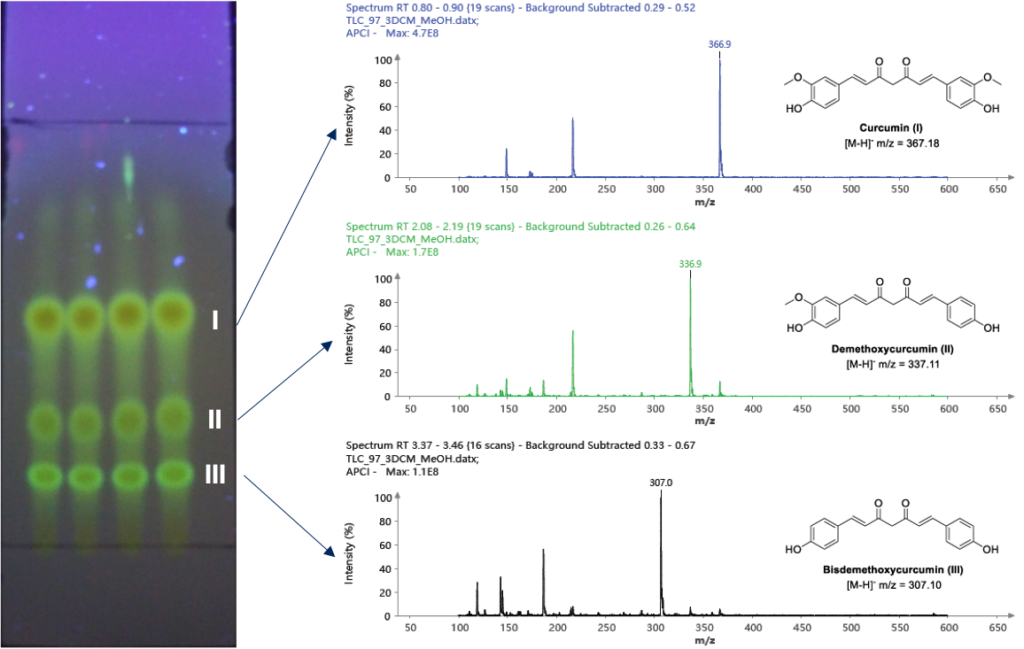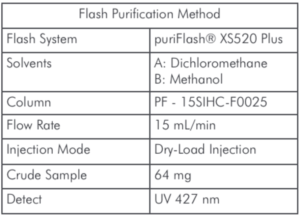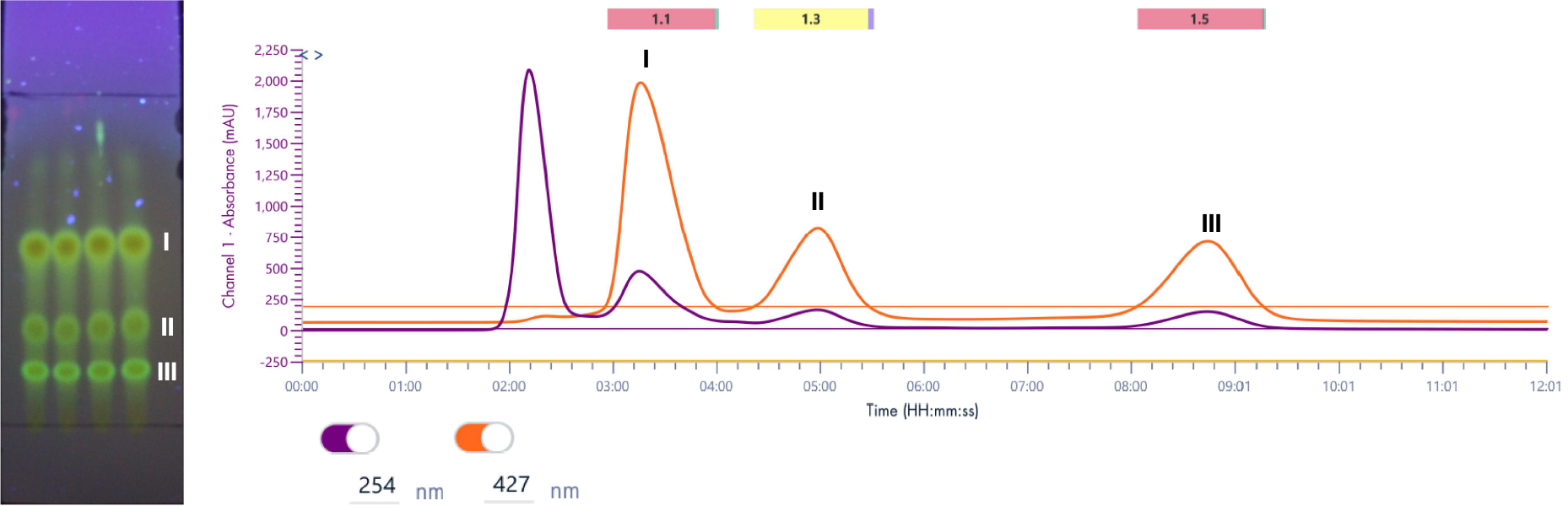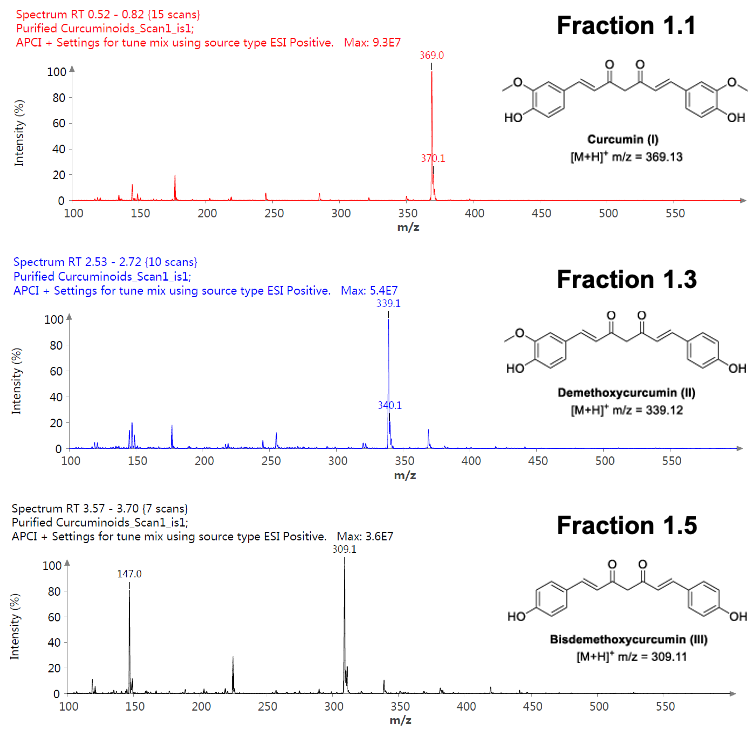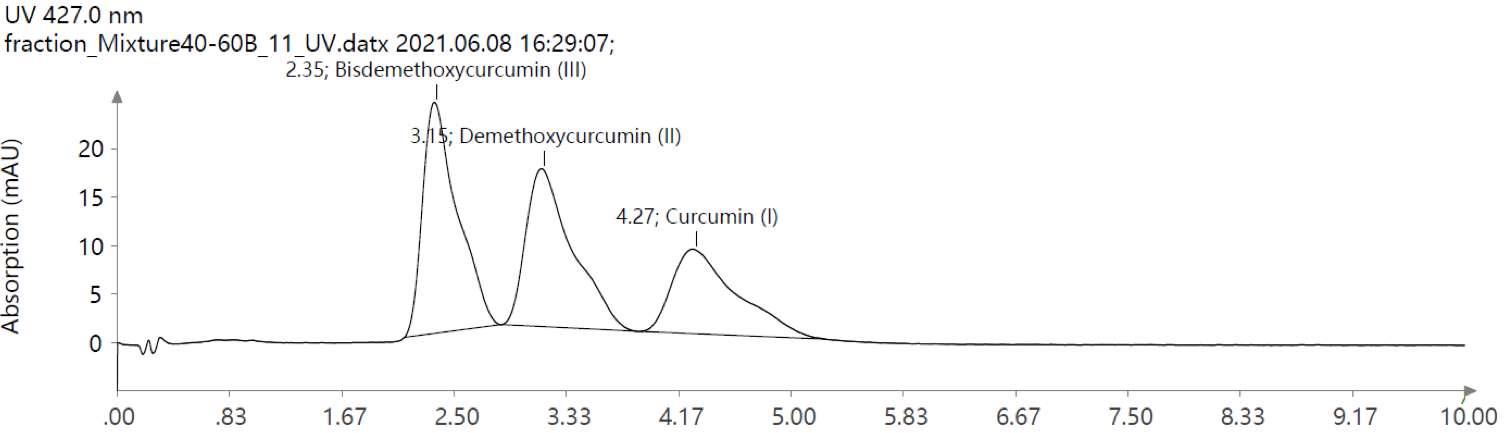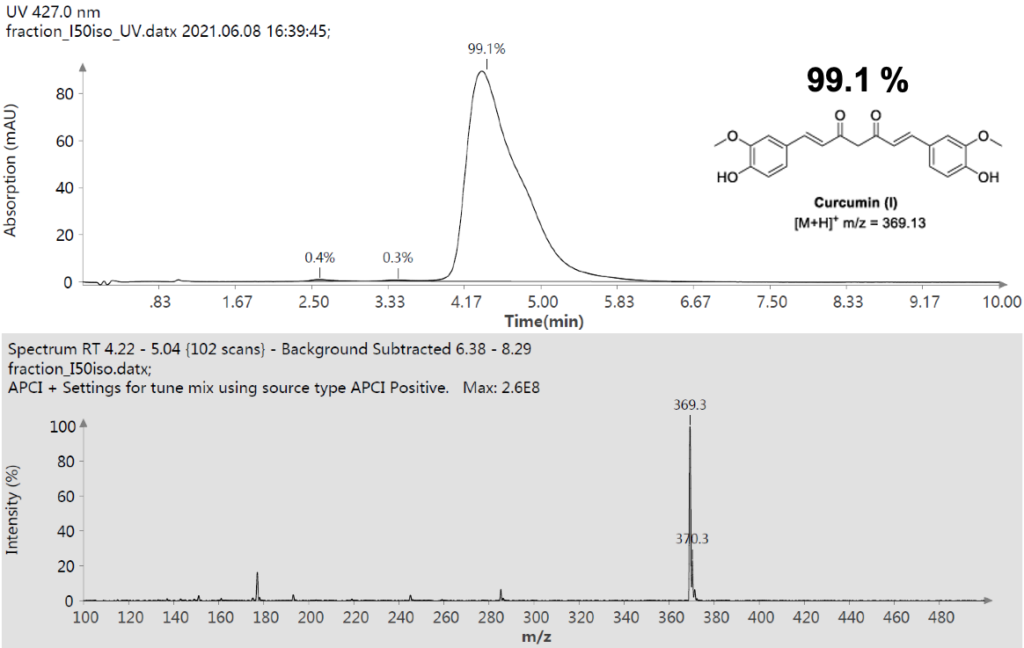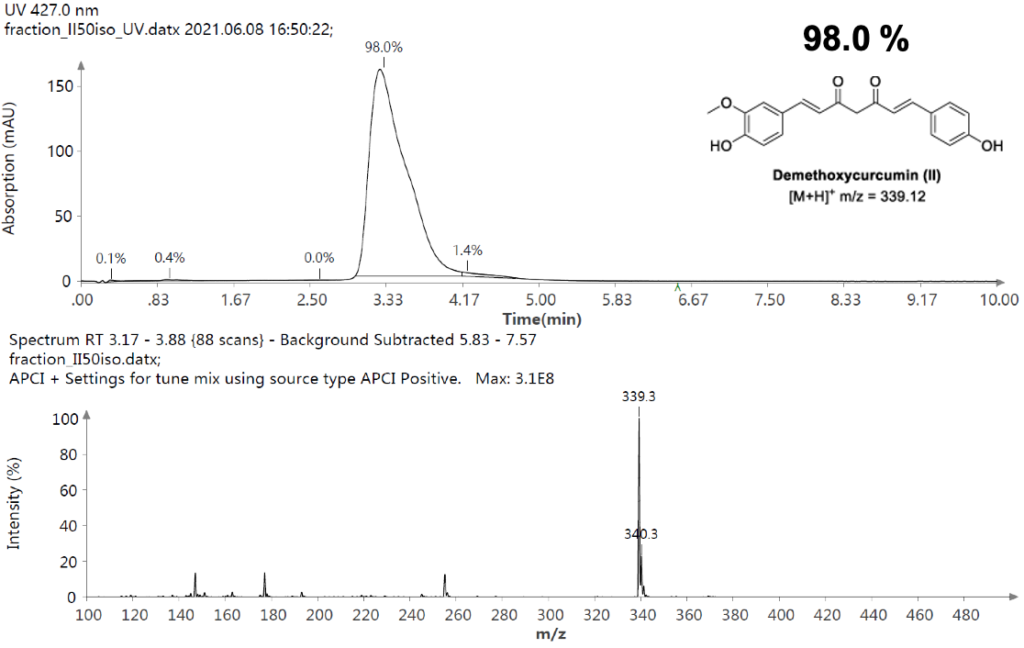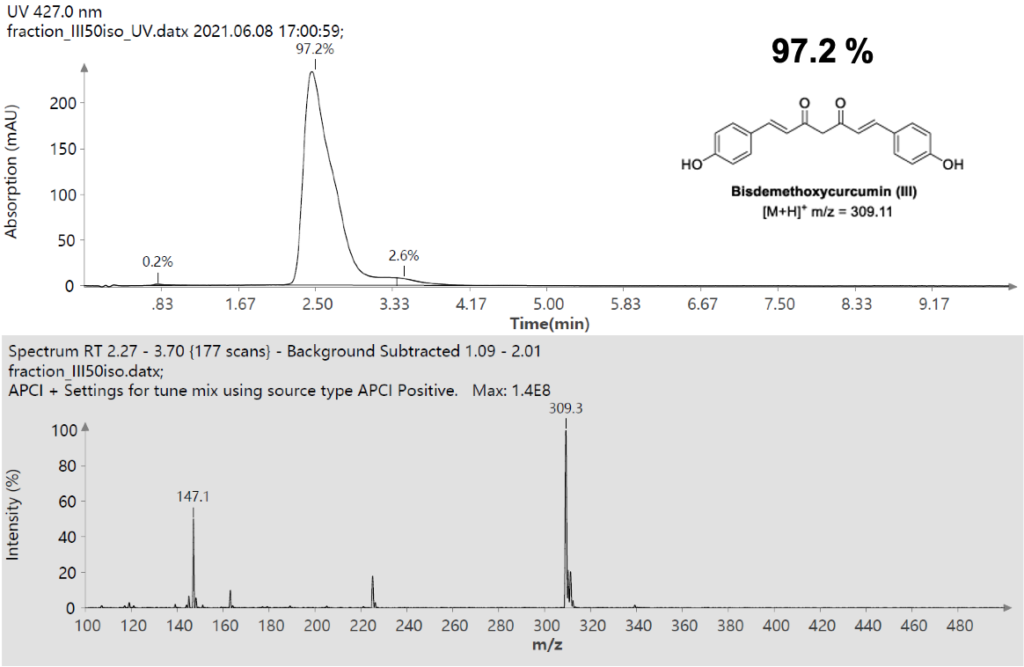
仪表
质谱:赛默飞世尔科技 Orbitrap® 仪器功能
打样TriVersa NanoMate LESA®
关键日期
乔什·库恩博士
威斯康星大学麦迪逊分校和 CeleramAb™
丹尼尔·艾克尔博士
Advion Interchim 科学®
引言
与小分子药物相比,生物制剂(或蛋白质疗法),例如抗体,由于其靶向生物通路具有更高的特异性,因此能够治疗癌症、自身免疫性疾病或代谢性疾病等复杂疾病,同时减少不良副作用,从而成为非常有效的药物。对这些生物制剂进行精确表征——包括其结构、翻译后修饰、稳定性和活性——对于确保其安全性、有效性和批次间一致性至关重要。即使是蛋白质折叠的微小变化,例如由单个氨基酸的化学修饰引起的变化,也可能降低蛋白质疗法的功能,甚至更糟的是,引发不良的免疫反应。因此,严格的分析方法能够指导最佳设计、生产工艺和质量控制。准确的表征还有助于获得监管部门的批准,并有助于识别能够改善治疗效果和患者预后的生物标志物或机制。
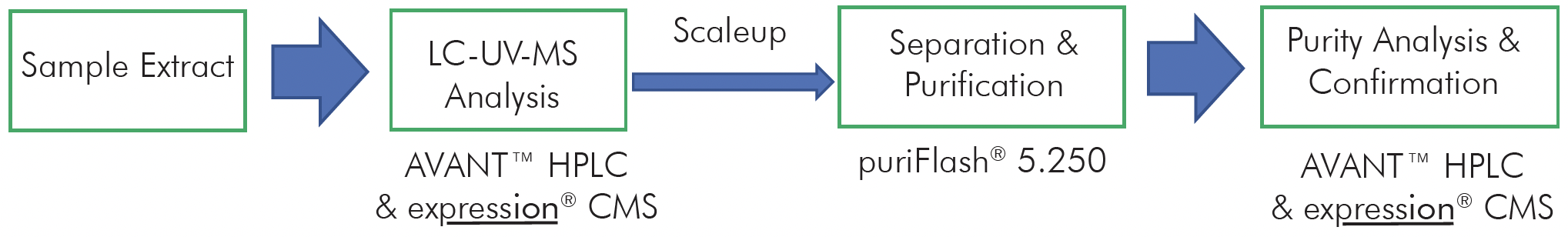
本文描述了一种基于96孔板标准化酶消化的蛋白质/抗体表征方法,该方法将生成的肽快速注入高分辨率质谱仪,并随后进行自动数据分析,以对每天多达1000个mAb进行完整的蛋白质序列表征(图1)。
Advion Interchim 科学® 系统


TriVersa NanoMate LESA® 带 ESI 芯片® 技术

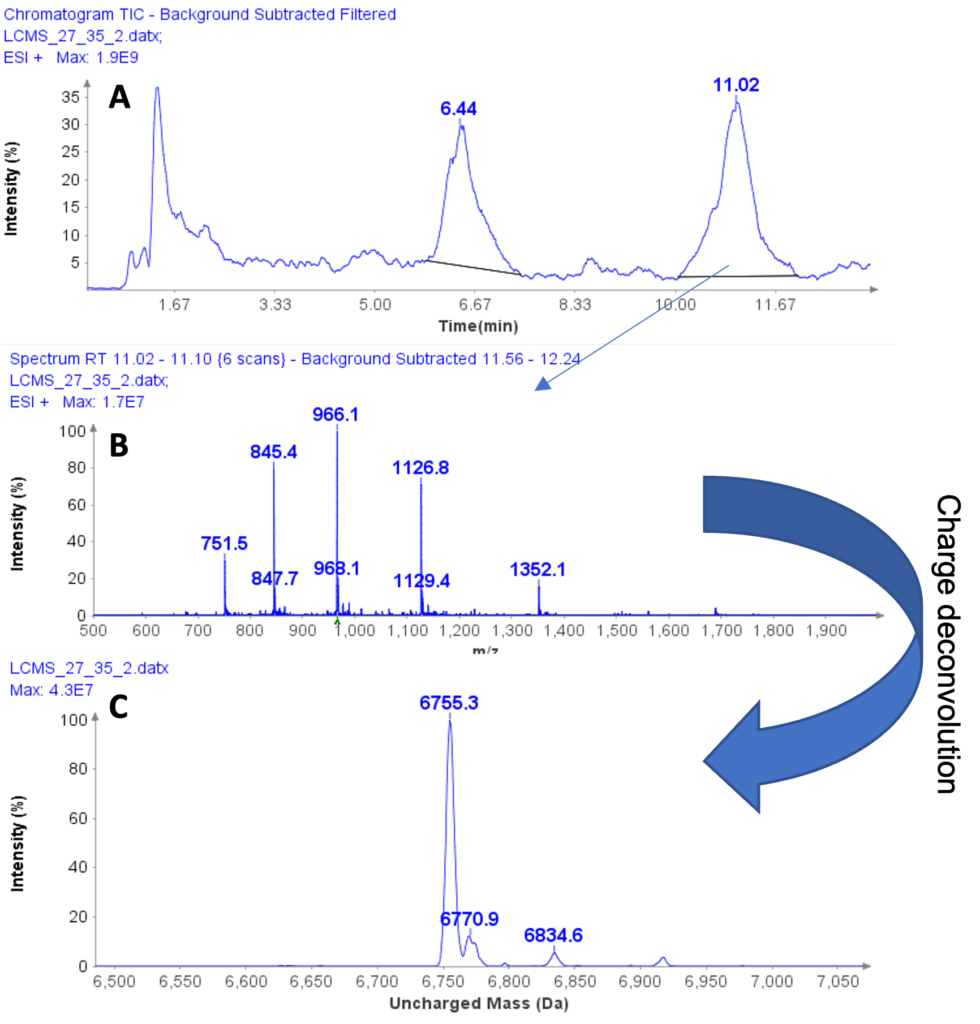
 图1结合 CeleramAb 的技术™ 以及 Advion Interchim Scientific® 创建基于酶解蛋白质、直接进样质谱、串联质谱和自动化数据处理的蛋白质治疗分析方法,以实现每天 1000 个 mAb 的通量。
图1结合 CeleramAb 的技术™ 以及 Advion Interchim Scientific® 创建基于酶解蛋白质、直接进样质谱、串联质谱和自动化数据处理的蛋白质治疗分析方法,以实现每天 1000 个 mAb 的通量。
概念与实验
蛋白质药物会因时间、pH值变化、光照或热暴露等多种因素而降解,导致生物药物肽链上的氨基酸发生改变,例如脱酰胺、氧化或热解,进而引起折叠和功能改变。图2展示了抗体上观察到的典型变化。糖基化、磷酸化或半胱氨酸-半胱氨酸氧化状态的其他变化也会影响生物制剂的折叠和功能。因此,所有这些修饰都必须进行研究、表征,并最终进行常规维护和控制,以开发出有价值的新药。
 图2蛋白质疗法会因时间、pH 值变化、光照或热暴露等各种因素而降解,导致生物药物肽序列中的氨基酸发生变化,如脱酰胺、氧化或热解。
图2蛋白质疗法会因时间、pH 值变化、光照或热暴露等各种因素而降解,导致生物药物肽序列中的氨基酸发生变化,如脱酰胺、氧化或热解。
分析蛋白质序列的典型方法是将蛋白质酶解成肽段,然后分别进行液相色谱-串联质谱(LC-MS/MS)分析。色谱分离肽段,质谱分析则对肽段进行质谱分析,生成碎片离子,从而获得序列信息。以往,LC-MS/MS分析耗时较长,通常需要30-120分钟,并且需要大量的离线数据分析才能获得完整的序列覆盖率和蛋白质表征。这两个因素严重限制了每天可处理的样品数量,并间接限制了治疗性蛋白质的实验数量。
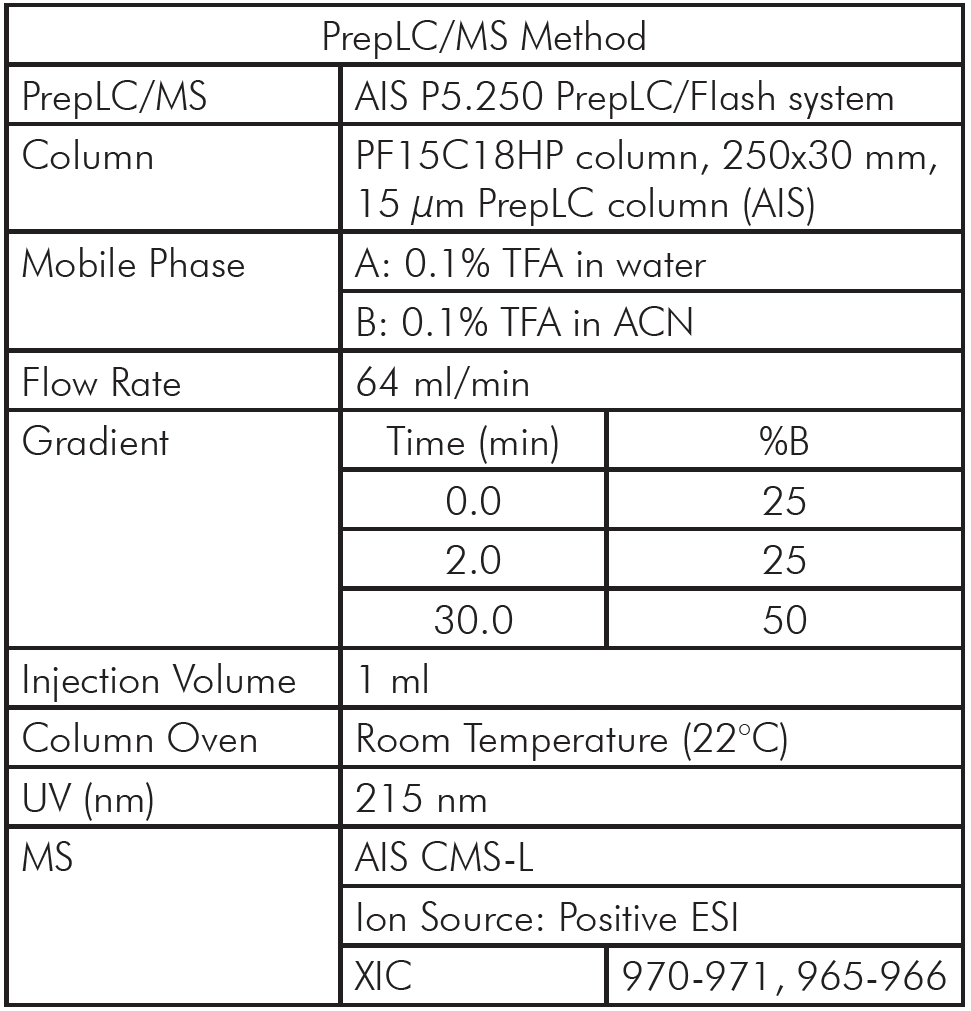
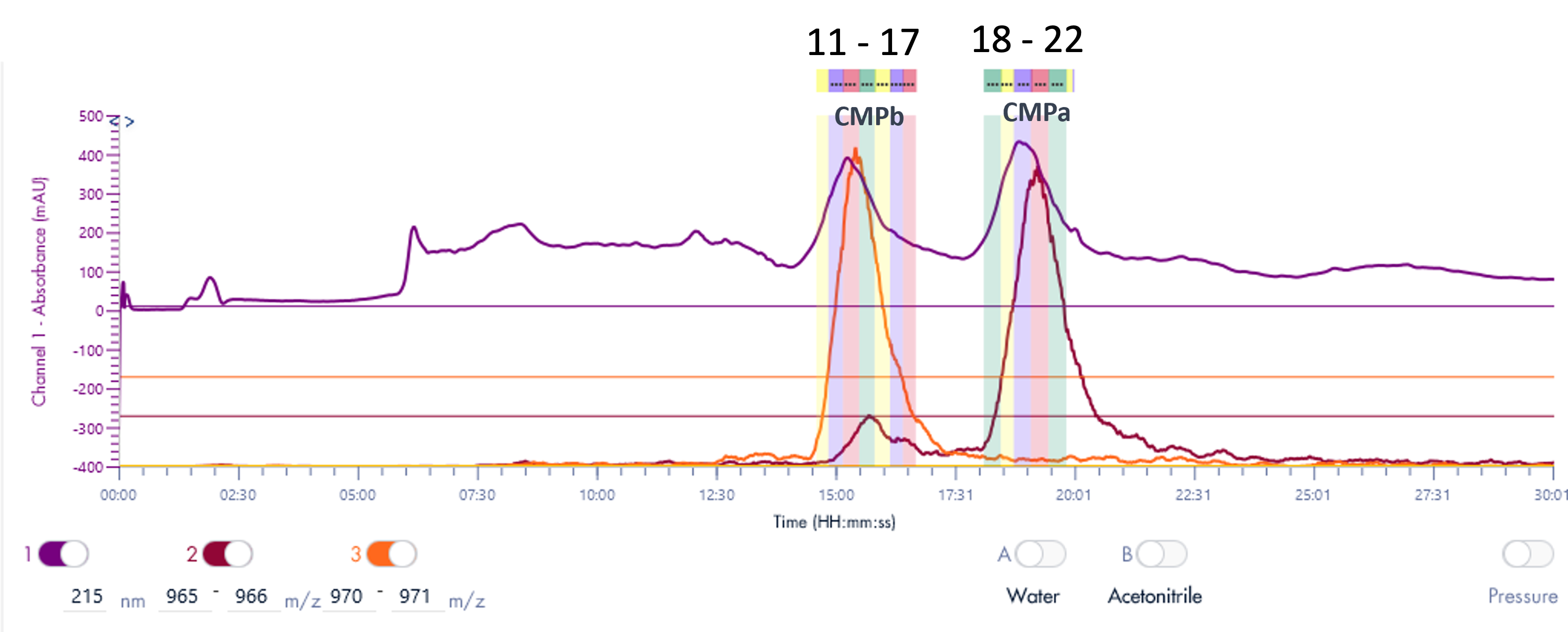
我们提出了一种自动化直接进样(DI)质谱分析方法,以替代耗时的液相色谱-串联质谱(LC-MS/MS)。由于现代质谱仪的MSn实验循环时间大幅缩短,并能实现高精度和高分辨率的质量分析,从而可以直接从质谱数据中明确鉴定肽序列及其修饰,因此该方法已成为可能。基于ESI chip®技术的自动化离子源的开发,使得每个连续样品仅需一个进样路径、一个进样头和一个新的nESI发射器,即可实现稳定高效的纳米电离(nano ESI),从而彻底消除交叉污染。如图3所示,该方法采用标准化的96孔板试剂,按照可重复的方案对抗体进行酶解,并使用NanoMate Triversa自动化机器人进样系统对生成的肽进行电离,然后在质谱仪中进行分析,仅需一分钟即可生成每个样品的信息丰富的质谱图。数据处理也通过定制软件实现自动化,以解决上述两个瓶颈问题,从而实现每天高达 1000 个样本的处理量。
 图3:肽图谱工作流程示意图,基于在 96 孔板中对蛋白质治疗药物进行标准化抗体消化,快速自动直接进样高分辨率质谱法生成信息丰富的 MS 数据(数据自动处理,每天可处理 1000 个 mAb)。
图3:肽图谱工作流程示意图,基于在 96 孔板中对蛋白质治疗药物进行标准化抗体消化,快速自动直接进样高分辨率质谱法生成信息丰富的 MS 数据(数据自动处理,每天可处理 1000 个 mAb)。
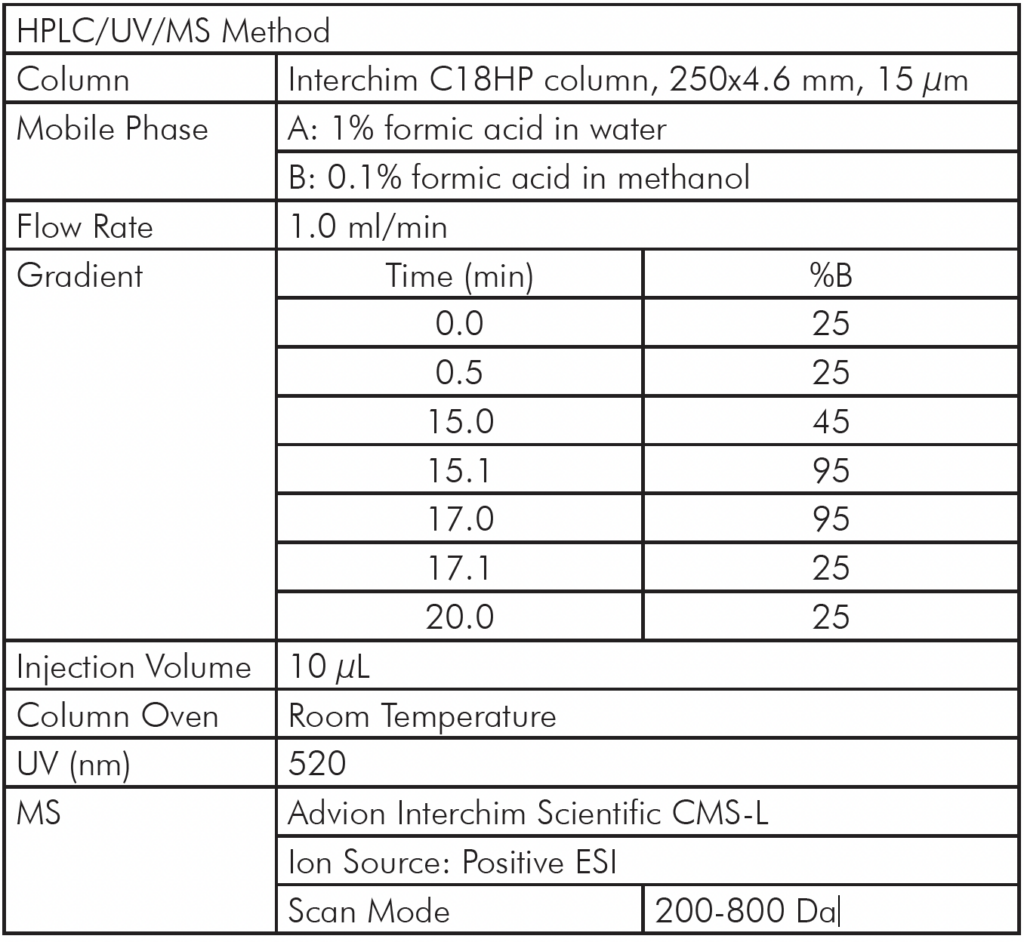
实验装置与方法
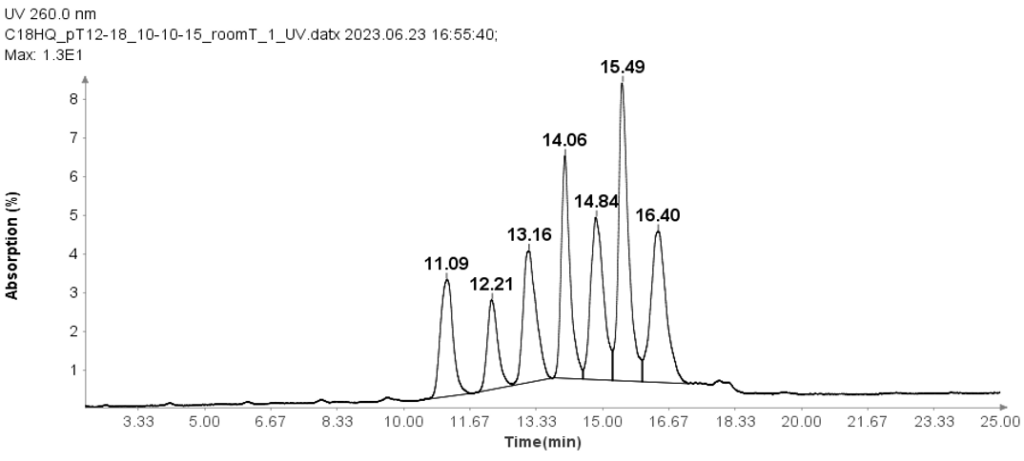
图 4 展示了采用直接进样-串联质谱 (DI-MS/MS) 方法进行的一组典型实验。其中,我们测试了两种不同的样品(抗体 X 和 NIST 标准抗体)在加热和 pH 值变化下的稳定性,每个样品均进行了三次重复实验。
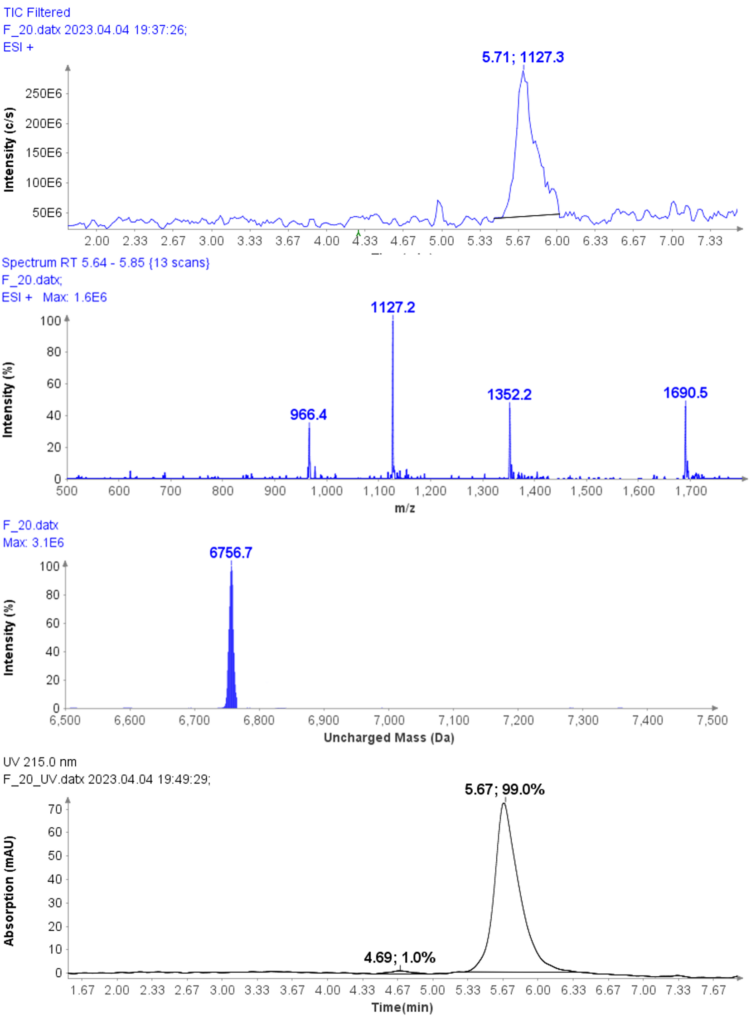
经过标准消化后,样品按所述方法注入质谱系统,并显示了原始质谱数据。在一个多小时内,分析了涵盖稳定性实验和对照的42个样品。在时间轴上,您可以看到每次1分钟进样实验的质谱数据强度峰值,随后是数据空白期(反映了机器人将下一个样品送入质谱系统所需的时间)。这一个小时与传统液相色谱-质谱联用方法分析单个样品所需的时间大致相同。
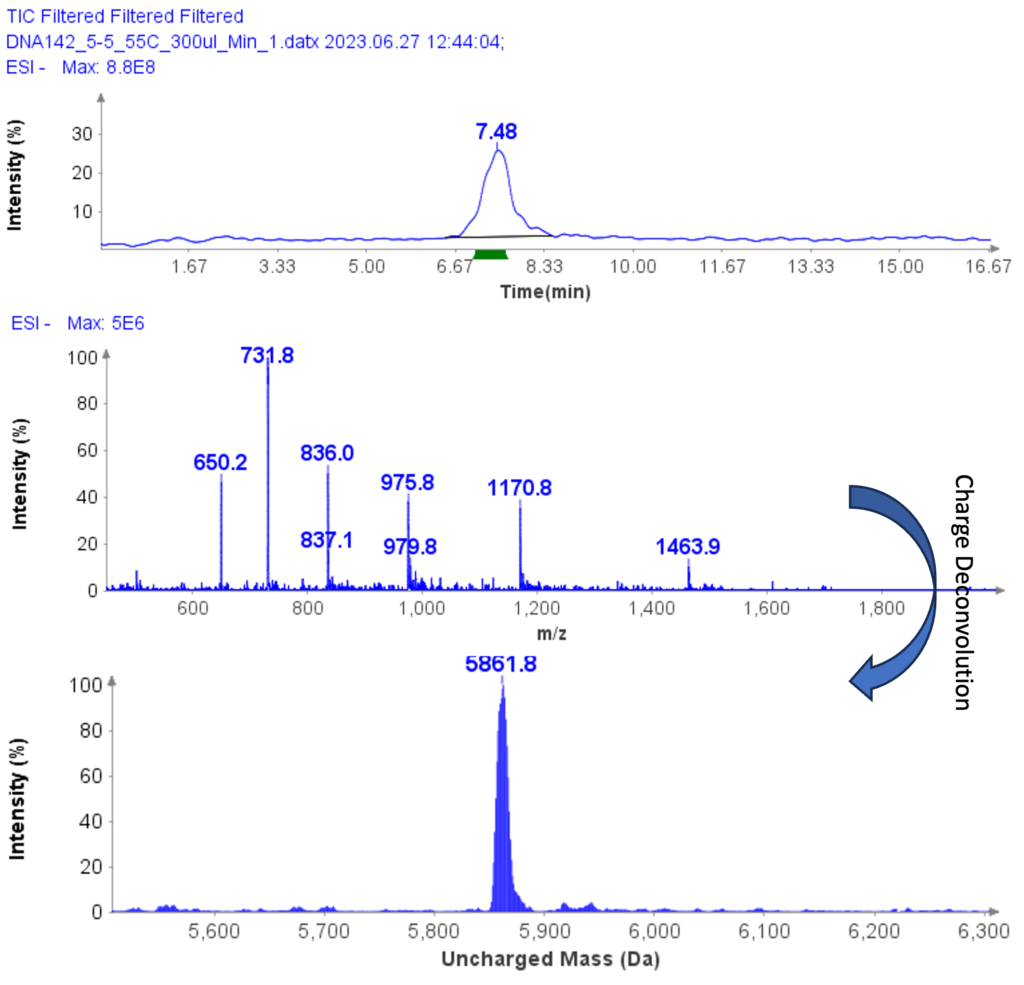
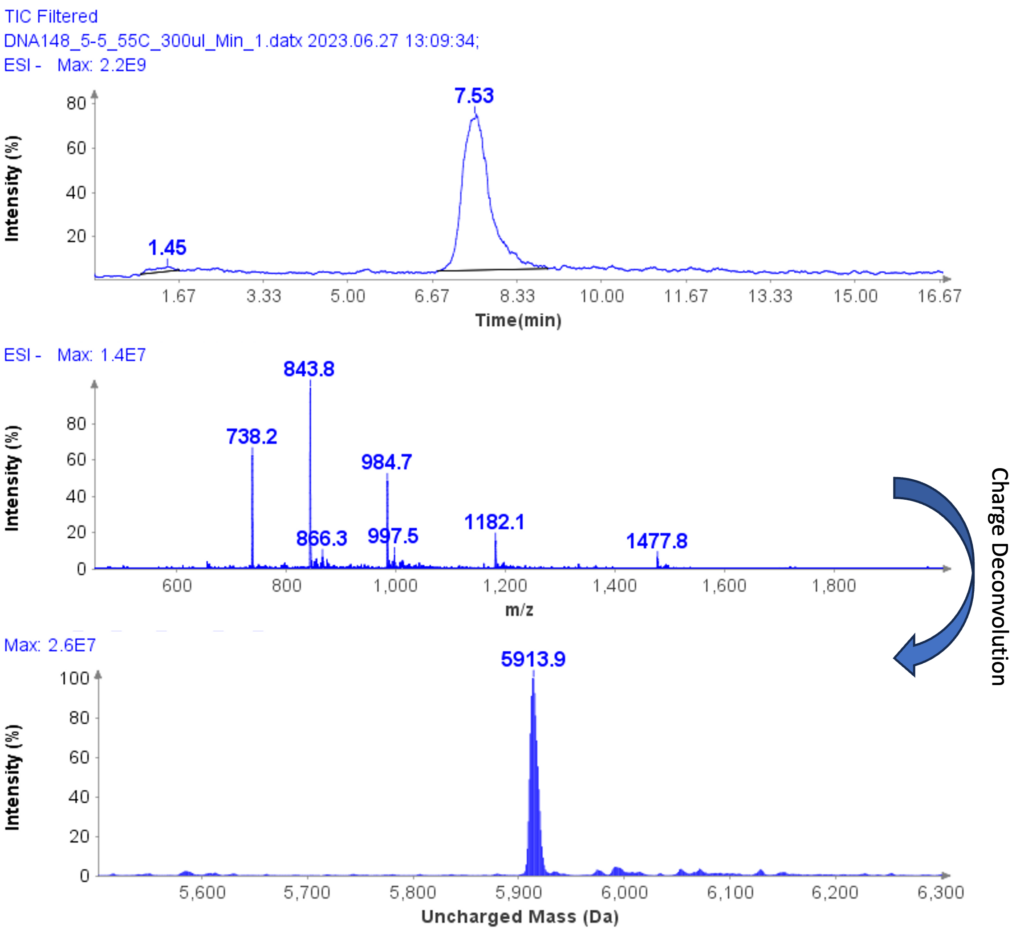
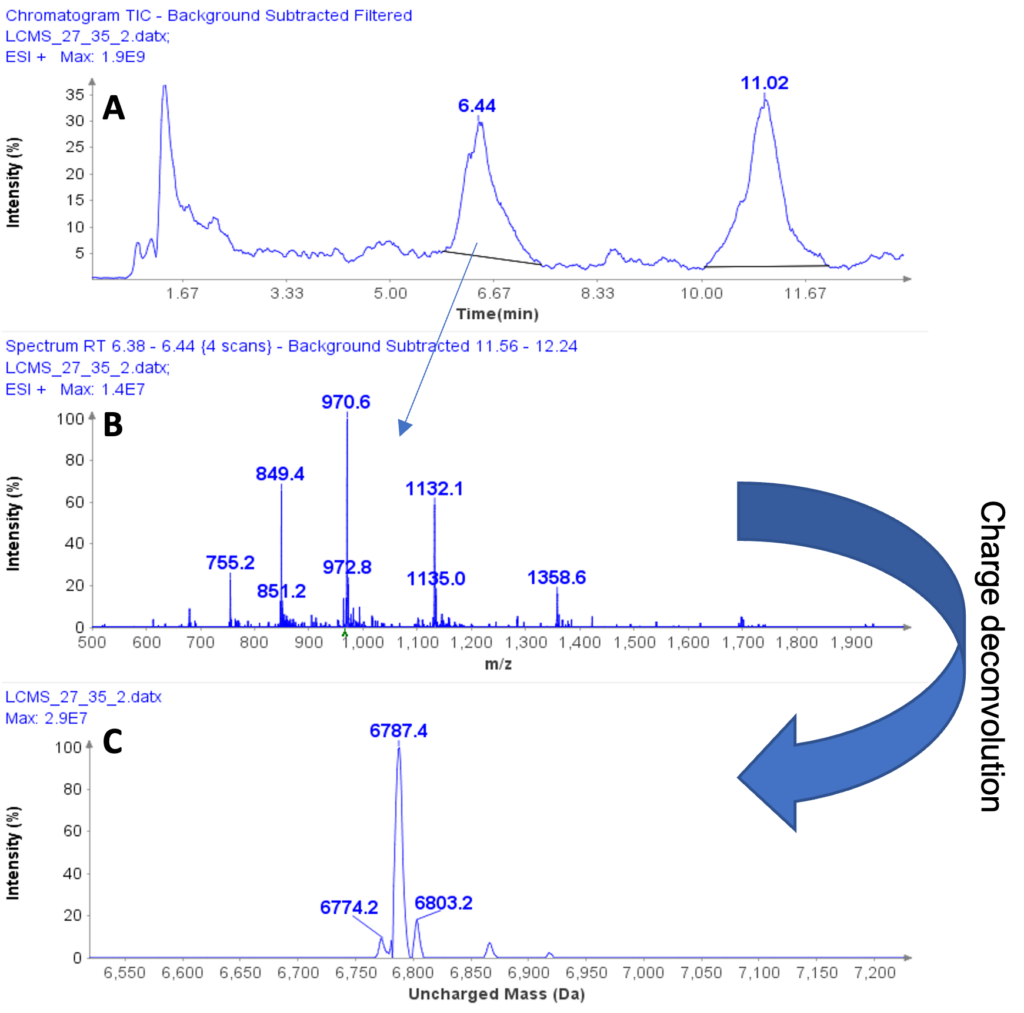
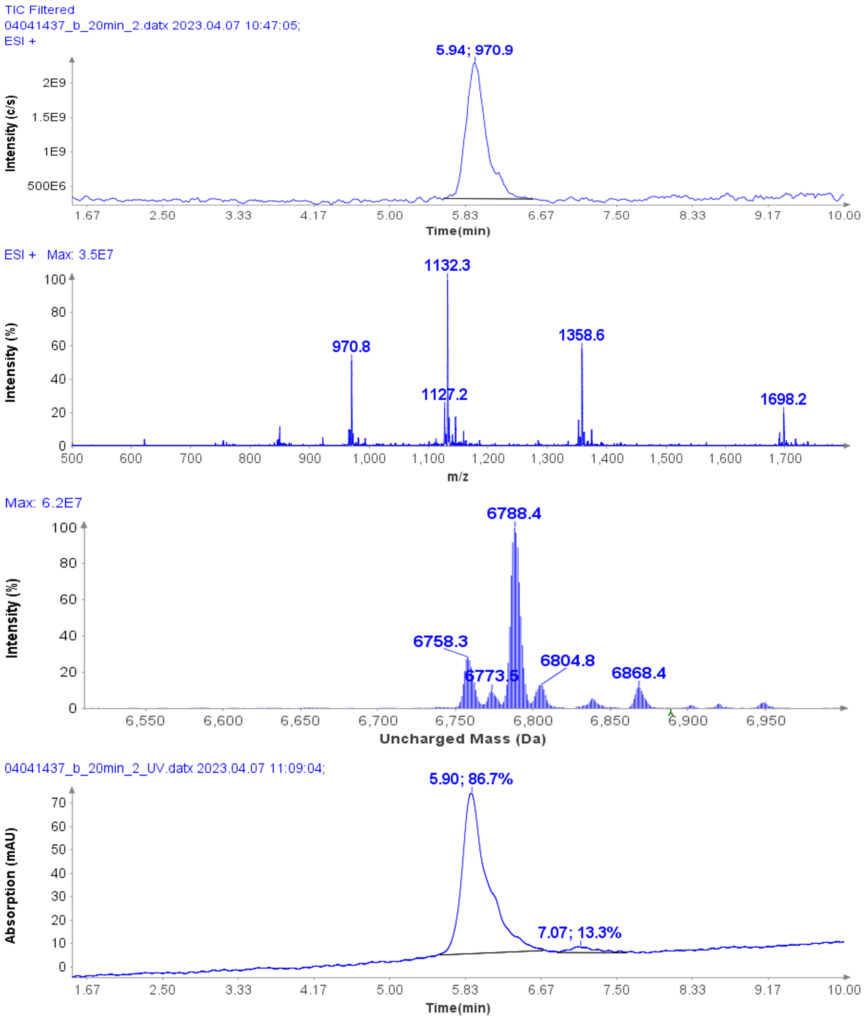
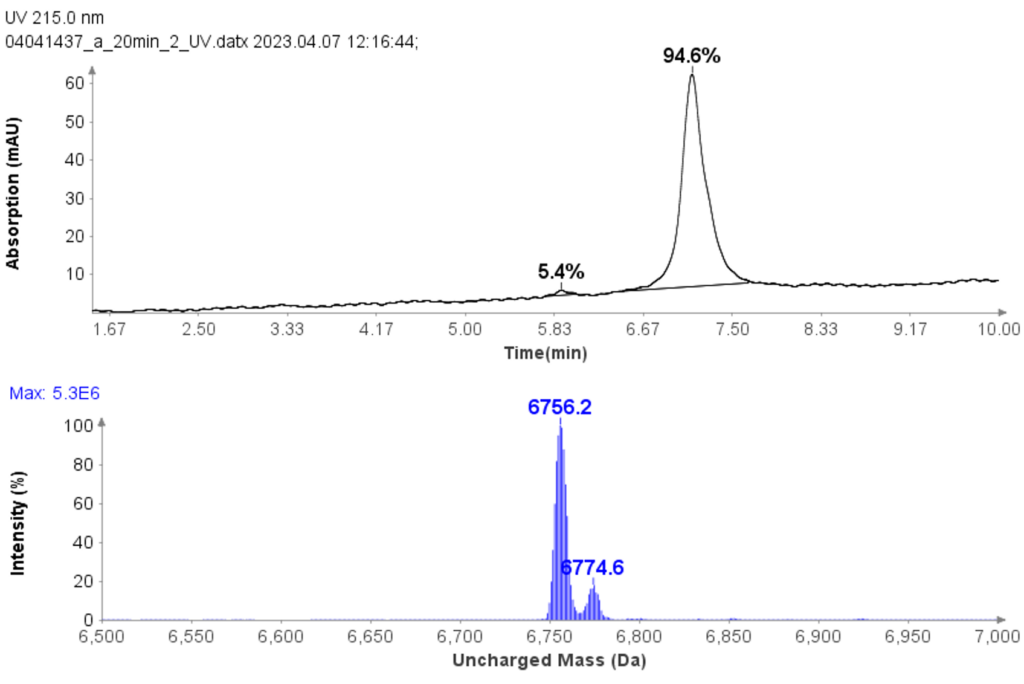
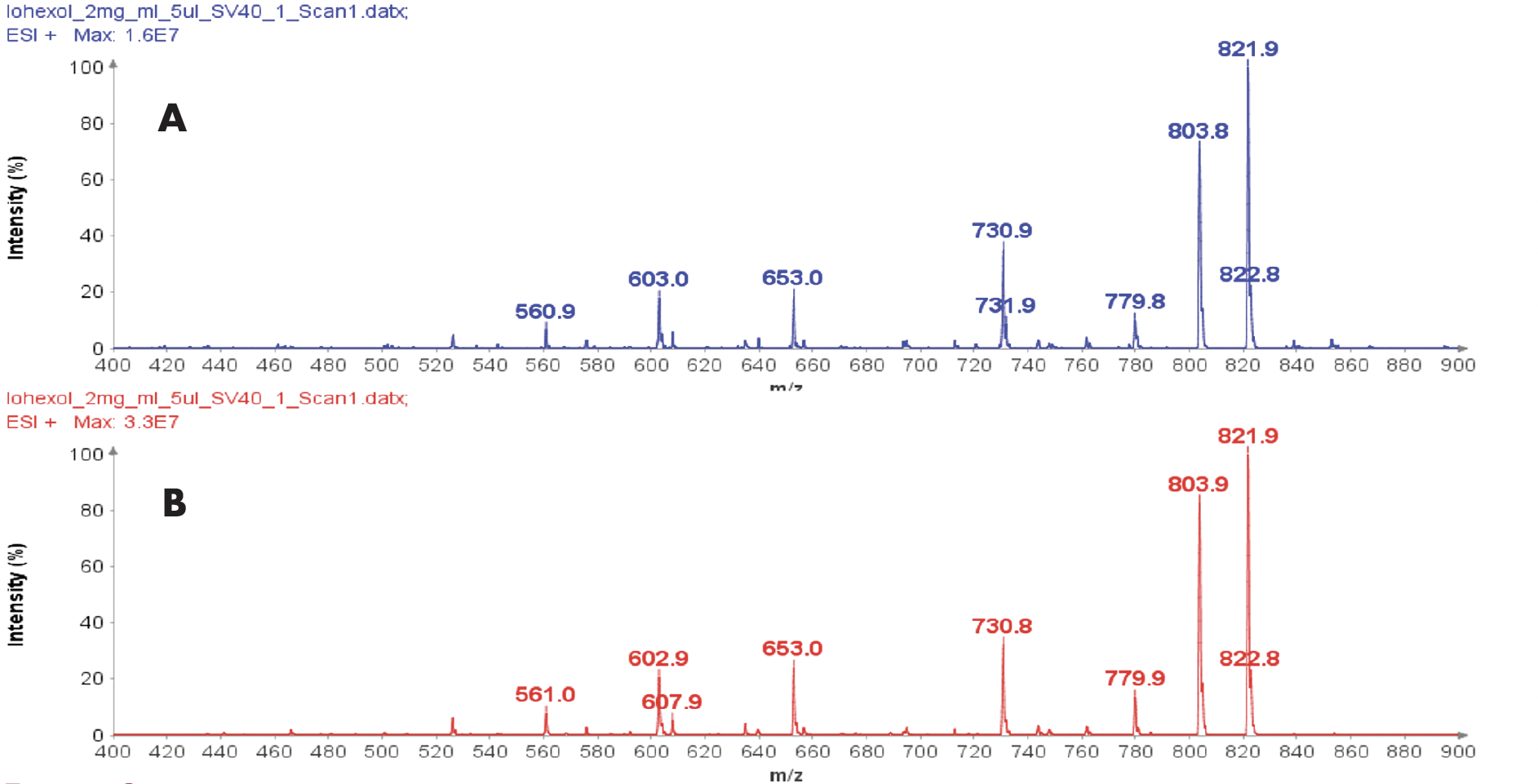
以肽段 DTLMISR 为例,展示了该工作流程。图 5 显示了治疗性抗体肽段序列 DTLM(ox)ISR 中氧化甲硫氨酸的质谱数据。典型的液相色谱-质谱 (LC-MS) 分析方法需要 30-120 分钟来分离蛋白质消化物,然后进行人工数据检查。然而,基于直接离子对串联质谱 (DI-MS/MS) 的方法仅需 1 分钟运行时间,即可在气相中通过质荷比分离两种相关肽段(天然态和氧化态),并通过自动质量偏移算法进行检测。氧化态的计算基于离子强度,结果为 19.6% 的氧化态,与 LC-MS 结果完全一致——然而,该方法仅需极短的时间即可获得所需信息。
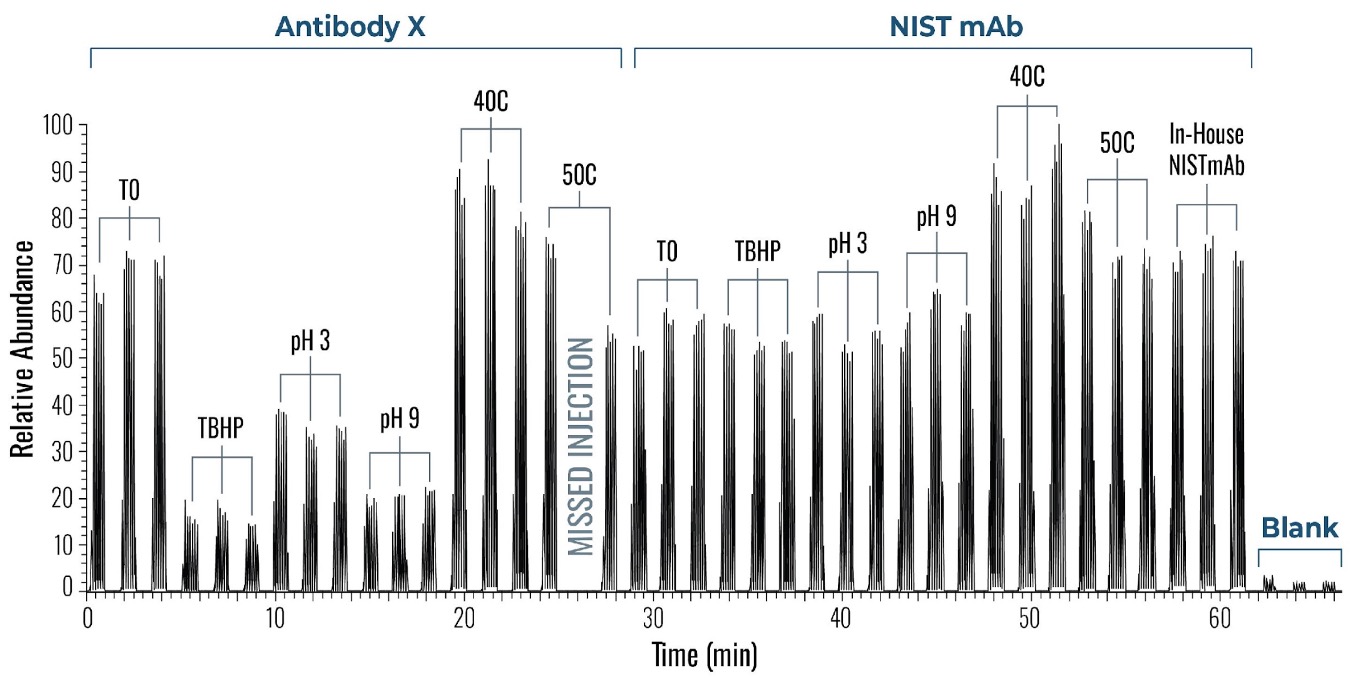 图4示例:一个包含 14 个样本的序列,每个样本重复三次,运行时间略超过一小时。两种蛋白质(抗体 X 和 NIST 标准单克隆抗体)均暴露于各种条件(pH 值和温度)下并进行测试。每次运行代表 1 分钟的进样 MS 数据,用于后续处理、肽段鉴定和修饰分析。
图4示例:一个包含 14 个样本的序列,每个样本重复三次,运行时间略超过一小时。两种蛋白质(抗体 X 和 NIST 标准单克隆抗体)均暴露于各种条件(pH 值和温度)下并进行测试。每次运行代表 1 分钟的进样 MS 数据,用于后续处理、肽段鉴定和修饰分析。
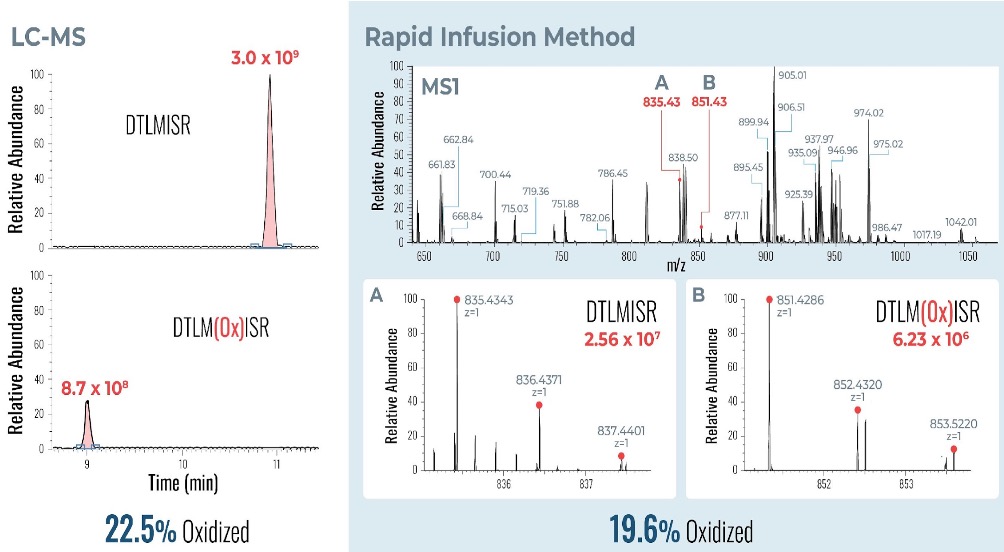

图5示例:对治疗性抗体肽序列 DTLMISR 中氧化甲硫氨酸的数据分析。典型的 LC-MS/MS 分析方法需要 30-120 分钟来分离蛋白质消化物,然后进行人工数据检查。然而,基于直接进样-MS/MS 的方法仅需 1 分钟运行时间,即可在气相中分离两个相关的肽序列,并通过自动质量偏移算法进行检测。氧化态的计算基于离子强度,结果为 19.6% 的氧化态,与 LC-MS/MS 结果完全一致——但所需时间却大大缩短。
结语
Advion Interchim 科学® TriVersa NanoMate® 自动化离子源是基于肽图谱策略表征治疗性蛋白的高通量工作流程的理想工具。与 CeleramAb 结合使用™ 通过标准化的试剂盒、质谱运行方法和自动化分析软件工具,我们可以实现通量比标准 LC-MS 方法提高 100 倍,一天最多可分析 1000 个 mAb。