Introduction
Agarwood is a highly valuable fragrant resinous wood which is widely used as traditional Chinese medicines, perfumes, incense and decorations. Due to its high economic value and excessive demand, this leads to a rising price and proliferation of fake commodities. Thus, strict authenticity identification and quality evaluation of agarwood are of great significance.
Objective
To establish a simple, rapid and non-destructive technique for identifying the authenticity of agarwood.
Methods
Liquid extraction surface analysis mass spectrometry (LESA-MS) was firstly proposed to identify the authenticity of 62 agarwood samples without sample preparation. In addition, multivariate statistical models and thin-layer chromatography (TLC) method were used to analyse and verify the results of LESA-MS.
Results
Representative compounds of agarwood were detected by LESA-MS. A characteristic 2-(2-phenylethyl)chromone compound (m/z 319.1) was treated as a key chemical marker to identify agarwood and its counterfeits rapidly. Several other chromones ions were identified and used as additional evidence for authentic samples. A total of 62 samples were visually discriminated as two groups by principal component analysis (PCA) and orthogonal projection to latent structures discriminant analysis (OPLS-DA), and the specific characteristic marker was highlighted. Moreover, the qualitative results of the conventional TLC method were in agreement with the LESA-MS approach.
Conclusion
The proposed LESA-MS method was successfully applied in the direct qualitative analysis of agarwood from different sources. This study indicated great feasibility and practicality of LESA-MS in the rapid identification of agarwood, and provided a non-destructive and meaningful preliminary screening tool for the agarwood industry.
Advion Interchim Scientific® Triversa NanoMate® (Advion, Ithaca, NY) was utilized.
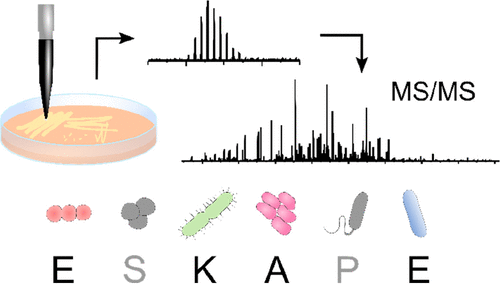
The ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae) represent clinically important bacterial species that are responsible for most hospital-acquired drug-resistant infections; hence, the need for rapid identification is of high importance. Previous work has demonstrated the suitability of liquid extraction surface analysis mass spectrometry (LESA-MS) for the direct analysis of colonies of two of the ESKAPE pathogens (Staphylococcus aureus and Pseudomonas aeruginosa) growing on agar. Here, we apply LESA-MS to the remaining four ESKAPE species (E. faecium E745, K. pneumoniae KP257, A. baumannii AYE, and E. cloacae S11) as well as E. faecalis V583 (a close relative of E. faecium) and a clinical isolate of A. baumannii AC02 using an optimized solvent sampling system. In each case, top-down LESA MS/MS was employed for protein identification. In total, 24 proteins were identified from 37 MS/MS spectra by searching against protein databases for the individual species. The MS/MS spectra for the identified proteins were subsequently searched against multiple databases from multiple species in an automated data analysis workflow with a view to determining the accuracy of identification of unknowns. Out of 24 proteins, 19 were correctly assigned at the protein and species level, corresponding to an identification success rate of 79%.
LESA-MS was performed using the Advion Interchim Scientific® TriVersa NanoMate®.
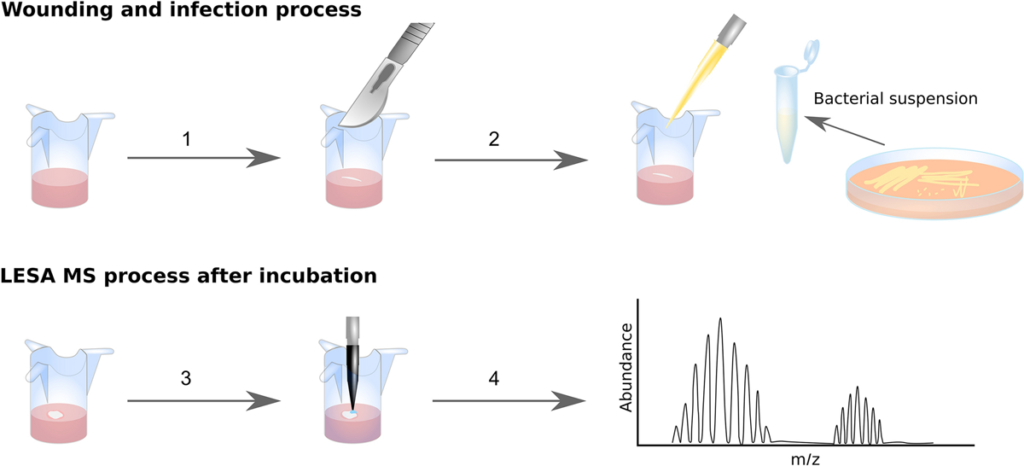
Trauma is one of the leading causes of death in people under the age of 49 and complications due to wound infection are the primary cause of death in the first few days after injury. The ESKAPE pathogens are a group of bacteria that are a leading cause of hospital-acquired infections and a major concern in terms of antibiotic resistance. Here, we demonstrate a novel and highly accurate approach for the rapid identification of ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) directly from infected wounds in 3D in vitro skin models. Wounded skin models were inoculated with bacteria and left to incubate. Bacterial proteins were identified within minutes, directly from the wound, by liquid extraction surface analysis mass spectrometry. This approach was able to distinguish closely related strains and, unlike genomic approaches, can be modified to provide dynamic information about pathogen behaviour at the wound site. In addition, since human skin proteins were also identified, this method offers the opportunity to analyse both host and pathogen biomarkers during wound infection in near real-time.
University of Cambridge, Royal Holloway University of London
Highlights
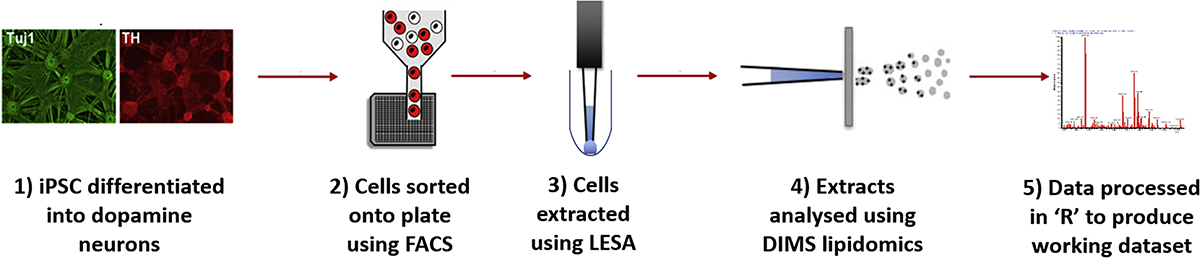
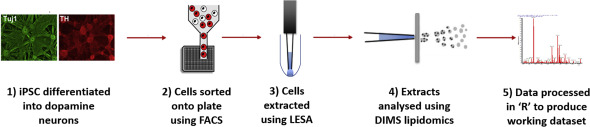
Combining FACS and LESA-MS to establish high-throughput single cell lipid profiling
Lipid differences found within and between populations of human dopamine neurons
Inter-cell lipid heterogeneity is increased in SNCA-A53T dopamine neurons
Identification and isolation of human iPSC-dopamine neurons with a TH-RFP reporter
Summary
Advances in single cell genomics and transcriptomics have shown that at tissue level there is complex cellular heterogeneity. To understand the effect of this inter-cell heterogeneity on metabolism, it is essential to develop a single cell lipid profiling approach that allows the measurement of lipids in large numbers of single cells from a population. This will provide a functional readout of cell activity and membrane structure. Using liquid extraction surface analysis coupled with high-resolution mass spectrometry we have developed a high-throughput method for untargeted single cell lipid profiling. This technological advance highlighted the importance of cellular heterogeneity in the functional metabolism of individual human dopamine neurons, suggesting that A53T alpha-synuclein (SNCA) mutant neurons have impaired membrane function. These results demonstrate that this single cell lipid profiling platform can provide robust data that will expand the frontiers in biomedical research.
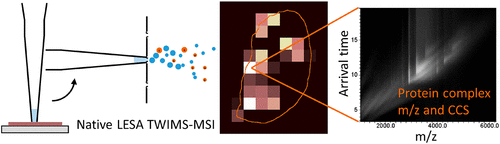
We have previously demonstrated native liquid extraction surface analysis (LESA) mass spectrometry imaging of small intact proteins in thin tissue sections. We also showed calculation of collision cross sections for specific proteins extracted from discrete locations in tissue by LESA traveling wave ion mobility spectrometry (TWIMS). Here, we demonstrate an integrated native LESA TWIMS mass spectrometry imaging (MSI) workflow, in which ion mobility separation is central to the imaging experiment and which provides spatial, conformational, and mass information on endogenous proteins in a single experiment. The approach was applied to MSI of a thin tissue section of mouse kidney. The results show that the benefits of integration of TWIMS include improved specificity of the ion images and the capacity to calculate collision cross sections for any protein or protein complex detected in any pixel (without a priori knowledge of the presence of the protein).
Authors: Cecilie Rosting, Jinglei Yu, and Helen J. Cooper*
Abstract
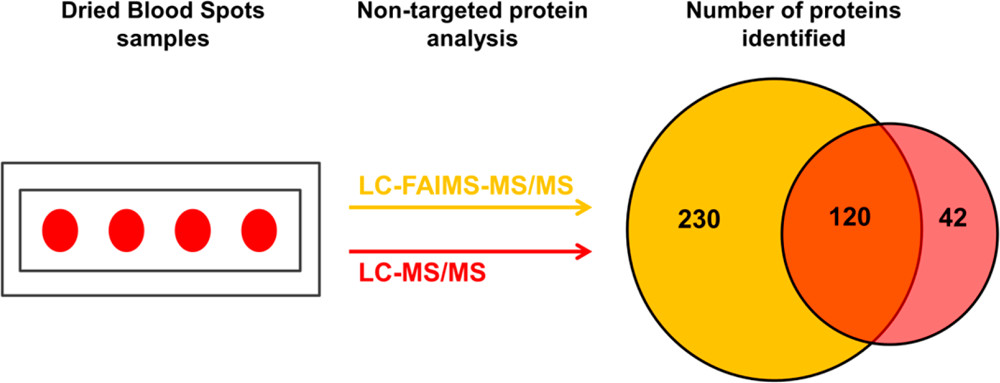
Despite the great potential of dried blood spots (DBS) as a source of endogenous proteins for biomarker discovery, the literature relating to nontargeted bottom-up proteomics of DBS is sparse, primarily due to the inherent complexity and very high dynamic range associated with these samples. Here, we present proof-of-concept results in which we have coupled high field asymmetric waveform ion mobility spectrometry (FAIMS) with liquid chromatography–tandem mass spectrometry (LC–MS/MS) for nontargeted bottom-up proteomics of DBS with the aim of addressing these challenges.
We, and others, have previously demonstrated the benefits of FAIMS more generally in proteomics including improved signal-to-noise and extended proteome coverage, and the aim of the current work was to extend those benefits specifically to DBS. The DBS samples were either extracted by the more traditional manual “punch and elute” approach or by an automated liquid surface extraction (LESA) approach prior to trypsin digestion. The resulting samples were analyzed by LC–MS/MS and LC–FAIMS–MS/MS analysis. The results show that the total number of proteins identified increased by ∼50% for the punch and elute samples and ∼45% for the LESA samples in the LC–FAIMS–MS/MS analysis. For both the punch and elute samples and the LESA samples, ∼30% of the total proteins identified were observed in both the LC–MS/MS and the LC–FAIMS–MS/MS data sets, illustrating the complementarity of the approaches.
Overall, this work demonstrates the benefits of inclusion of FAIMS for nontargeted proteomics of DBS.
Liquid extraction surface analysis (LESA) is an ambient surface sampling technique that allows the analysis of intact proteins directly from tissue samples via mass spectrometry. Integration of ion mobility separation to LESA mass spectrometry workflows has shown significant improvements in the signal-to-noise ratios of the resulting protein mass spectra and hence the number of proteins detected. Here, we report the use of a quadrupole–cyclic ion mobility–time-of-flight mass spectrometer (Q-cIM-ToF) for the analysis of proteins from mouse brain and rat kidney tissues sampled via LESA. Among other features, the instrument allows multiple pass cyclic ion mobility separation, with concomitant increase in resolving power. Single-pass experiments enabled the detection of 30 proteins from mouse brain tissue, rising to 44 when quadrupole isolation was employed. In the absence of ion mobility separation, 21 proteins were detected in rat kidney tissue including the abundant α- and β-globin chains from hemoglobin. Single-pass cyclic ion mobility mass spectrometry enabled the detection of 60 additional proteins. Multipass experiments of a narrow m/z range (m/z 870–920) resulted in the detection of 24 proteins (one pass), 37 proteins (two passes) and 54 proteins (three passes), thus demonstrating the benefits of improved mobility resolving power.
Helen J. Cooper, Emma K. Sisley, Jakub Ujma, Martin Palmer, Kevin Giles, Francisco A. Fernandez-Lima
Shanghai University of Traditional Chinese Medicine
Objective
To establish a simple, rapid and non‐destructive technique for identifying the authenticity of agarwood.
Methods
Liquid extraction surface analysis mass spectrometry (LESA‐MS) was firstly proposed to identify the authenticity of 62 agarwood samples without sample preparation. In addition, multivariate statistical models and thin‐layer chromatography (TLC) method were used to analyse and verify the results of LESA‐MS.
Conclusion
The proposed LESA‐MS method was successfully applied in the direct qualitative analysis of agarwood from different sources. This study indicated great feasibility and practicality of LESA‐MS in the rapid identification of agarwood, and provided a non‐destructive and meaningful preliminary screening tool for the agarwood industry.
The brain is a remarkably complex organ and cholesterol homeostasis underpins brain function. It is known that cholesterol is not evenly distributed across different brain regions; however, the precise map of cholesterol metabolism in the brain remains unclear. If cholesterol metabolism is to be correlated with brain function it is essential to generate such a map. Here we describe an advanced mass spectrometry platform to reveal spatial cholesterol metabolism in situ at 400-µm spot diameter on 10-µm tissue slices from mouse brain. We mapped, not only cholesterol, but also other biologically active sterols arising from cholesterol turnover in both wild type and mice lacking cholesterol 24S-hydroxylase (CYP46A1), the major cholesterol metabolizing enzyme.
Almost exclusively high throughput lipid profiling of samples from large epidemiological studies, where we study gene-lifestyle interactions. The method works with plasma samples as well as dried blood spots. The method is also applied to small-scale studies of specific disease groups, dietary interventions and model systems such as yeast. The TriVersa® NanoMate® is also used to study the lipid composition of tissues analyzed by LESA®.
Why did you incorporate the TriVersa® NanoMate® into your laboratory?
The TriVersa® NanoMate® is essential to efficiently analyze large-scale studies. It offers a really robust method for high throughput studies with minimal carryover.
Who would you recommend to purchase the TriVersa® NanoMate®?
I recommend the TriVersa® NanoMate® to laboratories with a large number of samples requiring a robust and reliable delivery system. The TriVersa® NanoMate® eliminates typical nanoelectrospray ionization challenges.
Do you have any publications or presentations using the TriVersa® NanoMate®?
Publication Highlight: Development and Application of High-Throughput Single Cell Lipid Profiling: A Study of SNCA-A53T Human Dopamine Neurons Snowden, et al. iScience, 2020, 23(10), 101703
Combining FACS and LESA-MS to establish high-throughput single cell lipid profiling. Research identifies lipid differences found within and between populations of human dopamine neurons.
Other Publications:
Snowden et al. Combining lipidomics and machine learning to measure clinical lipids in dried blood spots. Metabolomics. DOI: 10.1007/s11306-020-01703-0
Koulman et al. The development and validation of a fast and robust dried blood spot based lipid profiling method to study infant metabolism. Metabolomics. DOI: 10.1007/s11306-014-0628-z
Furse et al. A high-throughput platform for detailed lipidomic analysis of a range of mouse and human tissues. Analytical and Bioanalytical Chemistry. DOI: 10.1007/s00216-020-02511-0
Harshfield et al. An unbiased lipid phenotyping approach to study the genetic determinants of lipids and their associations with coronary heart disease risk factors. J Proteom Res. DOI: 10.1021/acs.jproteome.8b00786
Mann et al. Insights into genetic variants associated with NASH-fibrosis from metabolite profiling. Hum Mol Genet. DOI: 10.1093/hmg/ddaa162