Shaun N. Robertson, Fadi Soukarieh, Thomas M. White, Miguel Camara, Manuel Romero*, and Rian L. Griffiths*
Abstract
Previously, metabolites diffused or secreted from microbial samples have been analyzed via liquid chromatography–mass spectrometry (LC–MS) approaches following lengthy extraction protocols. Here, we present a model system for growing biofilms on discs before utilizing rapid and direct surface sampling MS, namely, liquid extraction surface analysis, to study the microbial exometabolome. One of the benefits of this approach is its surface-specific nature, enabling mimicking biofilm formation in a way that the study of planktonic liquid cultures cannot imitate. Even though Pseudomonas aeruginosa (P. aeruginosa), Staphylococcus aureus (S. aureus), and Candida albicans (C. albicans) have been studied previously in isolation, very few studies consider the complexity of the interplay between these pathogens, which are commonly combined causative agents of infection. Our model system provides a route to investigate changes in the exometabolome, such as metabolites that become circulatory in the presence of multiple pathogens. Our results agree with previous reports showing that 2-alkyl-4(1H)-quinolone signal molecules produced by P. aeruginosa are important markers of infection and suggest that methods for monitoring levels of 2-heptyl-4-hydroxyquinoline and 2,4-dihydroxyquinoline, as well as pyocyanin, could be beneficial in the determination of causative agents in interkingdom infection including P. aeruginosa. Furthermore, studying changes in exometabolome metabolites between pqs quorum sensing antagonists in treated and nontreated samples suggests suppression of phenazine production by P. aeruginosa. Hence, our model provides a rapid analytical approach to gaining a mechanistic understanding of bacterial signaling.
Advion Interchim Scientific® TriVersa NanoMate® (Advion Interchim Scientific®, Ithaca, NY) was utilized for the LESA sampling.
Q: What is the focus of your lab’s research? A: Our lab specializes in diagnostics and pathophysiology of rare genetic (metabolic) disease. Specifically, our work focuses on the development and introduction of untargeted metabolomics in diagnostics and on integrating genomics and metabolomics data to improve patient care. Together with our (inter)national collaborators we helped discover multiple novel genetic diseases in the past few years. The TriVersa NanoMate® is used to infuse metabolite extracts of patient or research samples onto a high resolution mass spectrometer.
Q: Why did you incorporate the TriVersa NanoMate® into your laboratory? A: The TriVersa NanoMate® provides an easy to use and efficient method for sample infusion. Furthermore, and essential for our diagnostic practice, it virtually eliminates the chances of carryover.
Q: Who would you recommend to purchase the TriVersa NanoMate®? A: I recommend the TriVersa NanoMate® to laboratories applying direct-infusion mass spectrometry for metabolomics (targeted and untargeted) analysis.
Q: Do you have any publications or presentations using the TriVersa NanoMate®? A: We performed untargeted metabolomics on dried blood spots of patients with 46 different genetic metabolic defects, and we simulated their whole-exome sequencing results in silico. We showed that for accurate prioritization of disease causing genes, it is essential to take into account not only the primary reaction of the affected protein but a larger network of potentially affected metabolites, multiple steps away from the primary reaction.
Cross-Omics: Integrating Genomics with Metabolomics in Clinical Diagnostics.
Kerkhofs MHPM, Haijes HA, Willemsen AM, van Gassen KLI, van der Ham M,
Gerrits J, de Sain-van der Velden MGM, Prinsen HCMT, van Deutekom HWM, van Hasselt PM, Verhoeven-Duif NM, Jans JJM. Metabolites. 2020 May 18;10(5):206.doi: 10.3390/metabo10050206.
Abstract
Lipidomics, defined as the large-scale study of the pathways and networks of cellular lipids in biological systems, is an emerging and rapidly expanding research field. Although lipidomics has only emerged as a distinct field within the past few years, numerous new discoveries and advances have already been made. Among them, multi- dimensional mass spectrometry (MS)-based shotgun lipidomics has distinguished itself as a robust and highly informative analytical platform for MS analyses of individual lipid molecular species directly from biological lipid extracts. The current platform for shotgun lipidomics includes a series of simple steps such as multiplexed extractions/reactions during sample preparation, intrasource separation/selective ionization, identification of individual lipid molecular species using building block-based multi-dimensional MS (MDMS), and quantitation of the identified individual lipid molecular species using a two-step ratiometric method.
Advion Interchim Scientific® TriVersa NanoMate® was featured in this publication.
Authors: Regensburg University Hospital, European Molecular Biology Laboratory, Germany; University of Southern Denmark, Denmark
Abstract
Lipidomics data require consideration of ions with near-identical masses, which comprises among others the Type-II isotopic overlap. This overlap occurs in series of lipid species differing only by number of double bonds (DBs) mainly because of the natural abundance of 13C-atoms. High-resolution mass spectrometry, such as Fourier-transform mass spectrometry (FTMS), is capable of resolving Type-II overlap depending on mass resolving power. In this work, we evaluated FTMS quantification accuracy of lipid species affected by Type-II overlap. Spike experiments with lipid species pairs of various lipid classes were analyzed by flow injection analysis-FTMS. Accuracy of quantification was evaluated without and with Type-II correction (using relative isotope abundance) as well as utilizing the first isotopic peak (M+1). Isobaric peaks, which were sufficiently resolved, were most accurate without Type-II correction. In cases of partially resolved peaks, we observed peak interference causing distortions in mass and intensity, which is a well-described phenomenon in FTMS. Concentrations of respective species were more accurate when calculated from M+1. Moreover, some minor species, affected by considerable Type-II overlap, could only be quantified by M+1. Unexpectedly, even completely unresolved peaks were substantially overcorrected by Type-II correction because of peak interference. The described method was validated including intraday and interday precisions for human serum and fibroblast samples. Taken together, our results show that accurate quantification of lipid species by FTMS requires resolution-depended data analysis.
Samples were infused using the Advion Interchim Scientific® TriVersa NanoMate®.
Authors: University of Graz, BioTechMed-Graz, Austria; Max Planck Institute of Molecular Cell Biology and Genetics, Germany
Abstract
Triacylglycerol (TG) and steryl ester (SE) lipid storage is a universal strategy to maintain organismal energy and membrane homeostasis. Cycles of building and mobilizing storage fat are fundamental in (re)distributing lipid substrates between tissues or to progress ontogenetic transitions. In this study, we show that Hormone-sensitive lipase (Hsl) specifically controls SE mobilization to initiate intergenerational sterol transfer in Drosophila melanogaster. Tissue-autonomous Hsl functions in the maternal fat body and germline coordinately prevent adult SE overstorage and maximize sterol allocation to embryos. While Hsl-deficiency is largely dispensable for normal development on sterol-rich diets, animals depend on adipocyte Hsl for optimal fecundity when dietary sterol becomes limiting. Notably, accumulation of SE but not of TG is a characteristic of Hsl-deficient cells across phyla including murine white adipocytes. In summary, we identified Hsl as an ancestral regulator of SE degradation, which improves intergenerational sterol transfer and reproductive success in flies.
Nano-ESI analysis was performed by chip-based infusion using the Advion Interchim Scientific® TriVersa NanoMate®.
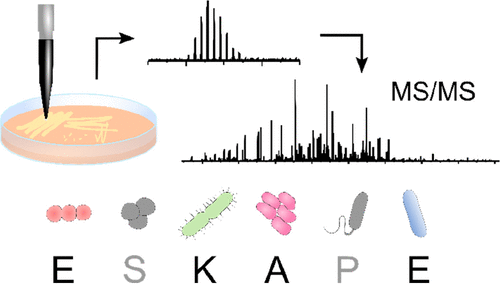
The ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae) represent clinically important bacterial species that are responsible for most hospital-acquired drug-resistant infections; hence, the need for rapid identification is of high importance. Previous work has demonstrated the suitability of liquid extraction surface analysis mass spectrometry (LESA-MS) for the direct analysis of colonies of two of the ESKAPE pathogens (Staphylococcus aureus and Pseudomonas aeruginosa) growing on agar. Here, we apply LESA-MS to the remaining four ESKAPE species (E. faecium E745, K. pneumoniae KP257, A. baumannii AYE, and E. cloacae S11) as well as E. faecalis V583 (a close relative of E. faecium) and a clinical isolate of A. baumannii AC02 using an optimized solvent sampling system. In each case, top-down LESA MS/MS was employed for protein identification. In total, 24 proteins were identified from 37 MS/MS spectra by searching against protein databases for the individual species. The MS/MS spectra for the identified proteins were subsequently searched against multiple databases from multiple species in an automated data analysis workflow with a view to determining the accuracy of identification of unknowns. Out of 24 proteins, 19 were correctly assigned at the protein and species level, corresponding to an identification success rate of 79%.
LESA-MS was performed using the Advion Interchim Scientific® TriVersa NanoMate®.
Authors: Heinrich Pette Institute – Leibniz Institute for Experimental Virology, European XFEL GmbH, Vienna University of Technology (TU Wien), Indiana University, University Medical Center Hamburg-Eppendorf, Bernard Nocht Institute for Tropical Medicine and German Center for Infection Research
Abstract
Noroviruses cause immense sporadic gastroenteritis outbreaks worldwide. Emerging genotypes, which are divided based on the sequence of the major capsid protein VP1, further enhance this public threat. Self-assembling properties of the human norovirus major capsid protein VP1 are crucial for using virus-like particles (VLPs) for vaccine development. However, there is no vaccine available yet. Here, VLPs from different variants produced in insect cells were characterized in detail using a set of biophysical and structural tools. We used native mass spectrometry, gas-phase electrophoretic mobility molecular analysis, and proteomics to get clear insights into particle size, structure, and composition, as well as stability. Generally, noroviruses have been known to form mainly T = 3 particles. Importantly, we identified a major truncation in the capsid proteins as a likely cause for the formation of T = 1 particles. For vaccine development, particle production needs to be a reproducible, reliable process. Understanding the underlying processes in capsid size variation will help to produce particles of a defined capsid size presenting antigens consistent with intact virions. Next to vaccine production itself, this would be immensely beneficial for bio-/nano-technological approaches using viral particles as carriers or triggers for immunological reactions.
This publication features the Advion TriVersa NanoMate® as an automated nano-ESI source for CDMS measurements.
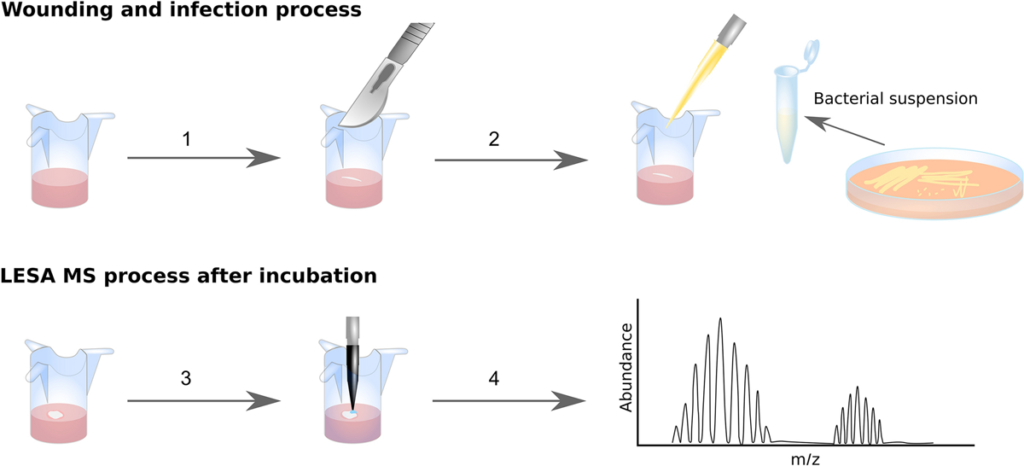
Trauma is one of the leading causes of death in people under the age of 49 and complications due to wound infection are the primary cause of death in the first few days after injury. The ESKAPE pathogens are a group of bacteria that are a leading cause of hospital-acquired infections and a major concern in terms of antibiotic resistance. Here, we demonstrate a novel and highly accurate approach for the rapid identification of ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) directly from infected wounds in 3D in vitro skin models. Wounded skin models were inoculated with bacteria and left to incubate. Bacterial proteins were identified within minutes, directly from the wound, by liquid extraction surface analysis mass spectrometry. This approach was able to distinguish closely related strains and, unlike genomic approaches, can be modified to provide dynamic information about pathogen behaviour at the wound site. In addition, since human skin proteins were also identified, this method offers the opportunity to analyse both host and pathogen biomarkers during wound infection in near real-time.
University of Cambridge, Royal Holloway University of London
Highlights
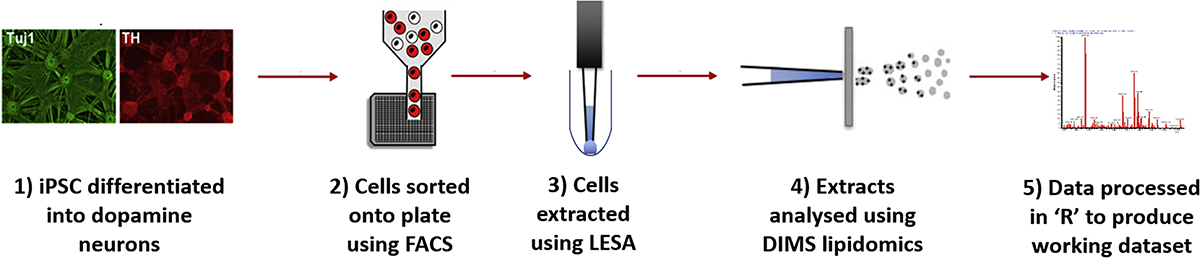
Combining FACS and LESA-MS to establish high-throughput single cell lipid profiling
Lipid differences found within and between populations of human dopamine neurons
Inter-cell lipid heterogeneity is increased in SNCA-A53T dopamine neurons
Identification and isolation of human iPSC-dopamine neurons with a TH-RFP reporter
Summary
Advances in single cell genomics and transcriptomics have shown that at tissue level there is complex cellular heterogeneity. To understand the effect of this inter-cell heterogeneity on metabolism, it is essential to develop a single cell lipid profiling approach that allows the measurement of lipids in large numbers of single cells from a population. This will provide a functional readout of cell activity and membrane structure. Using liquid extraction surface analysis coupled with high-resolution mass spectrometry we have developed a high-throughput method for untargeted single cell lipid profiling. This technological advance highlighted the importance of cellular heterogeneity in the functional metabolism of individual human dopamine neurons, suggesting that A53T alpha-synuclein (SNCA) mutant neurons have impaired membrane function. These results demonstrate that this single cell lipid profiling platform can provide robust data that will expand the frontiers in biomedical research.
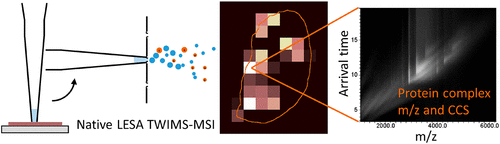
We have previously demonstrated native liquid extraction surface analysis (LESA) mass spectrometry imaging of small intact proteins in thin tissue sections. We also showed calculation of collision cross sections for specific proteins extracted from discrete locations in tissue by LESA traveling wave ion mobility spectrometry (TWIMS). Here, we demonstrate an integrated native LESA TWIMS mass spectrometry imaging (MSI) workflow, in which ion mobility separation is central to the imaging experiment and which provides spatial, conformational, and mass information on endogenous proteins in a single experiment. The approach was applied to MSI of a thin tissue section of mouse kidney. The results show that the benefits of integration of TWIMS include improved specificity of the ion images and the capacity to calculate collision cross sections for any protein or protein complex detected in any pixel (without a priori knowledge of the presence of the protein).