Abstract
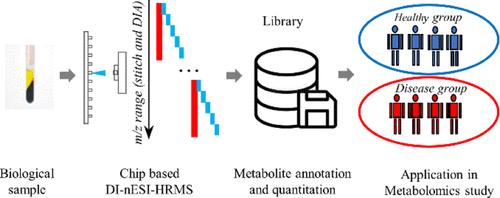
Direct-infusion nanoelectrospray ionization high-resolution mass spectrometry (DI-nESI-HRMS) is an alternative approach to chromatography–MS-based techniques for nontargeted metabolomics, offering a high sample throughout. However, its annotation accuracy of analytes is still full of challenges. In this study, we proposed a strategy for the annotation and quantitation of nontargeted metabolomic data using a spectral-stitching DI-nESI-HRMS with data-independent acquisition. The metabolite annotation strategy included the isotopic distribution, MS/MS spectrum similarity, and precursor and product ion correlation as well as matching of the extracted metabolite features along with the targeted metabolite precursors. Two groups of mixed standard solutions containing 40 and 79 metabolites were, respectively, used to establish the metabolite annotation strategy and validate its reliability. The results showed that the detected standards could be well annotated at top three explanations and total qualitative percentages were 100% (40 of 40) for the standard solution and 94.9% (74 of 78) for the standards spiked into the serum matrix. The intensity of the precursor ions was used for quantitation except for isomers, which were quantified by the intensities of the characteristic product ions if available. Finally, the strategy was applied to study serum metabolomics in diabetes, and the results demonstrated that it is promising for a large-scale cohort metabolomic study.
Advion Interchim Scientific® Triversa NanoMate® (Advion, Ithaca, NY) was utilized.
Abstract
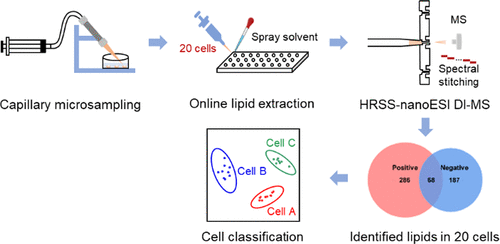
Studies of cellular metabolism can provide profound insights into the underlying molecular mechanisms and metabolic function. To date, the majority of cellular metabolism studies based on chromatography–mass spectrometry (MS) require population cells to obtain informative metabolome. These methods are not only time-consuming but also not suitable for amount-limited cell samples such as circulating tumor cells, stem cells, and neurons. Therefore, it is extremely essential to develop analytical methods enabling to detect metabolome from tens of cells in a high-throughput and high-sensitivity way. In this work, a novel platform for rapid and sensitive detection of lipidome in 20 mammalian cells was proposed using capillary microsampling combined with high-resolution spectral stitching nanoelectrospray ionization direct-infusion MS. It can be used to collect cells rapidly and accurately via the capillary microprobe, extract lipids directly in a 96-well plate using a spray solvent, and detect more than 500 lipids covering 19 lipid subclasses within 3 min. This novel platform was successfully applied to study the lipid features of different cancer cell types and subtypes as well as target cells from tissue samples. This study provides a strategy for determining the lipid species with rich information in tens of cells and demonstrates great potential for clinical applications.
Advion Interchim Scientific® Triversa NanoMate® (Advion, Ithaca, NY) was utilized.
Abstract
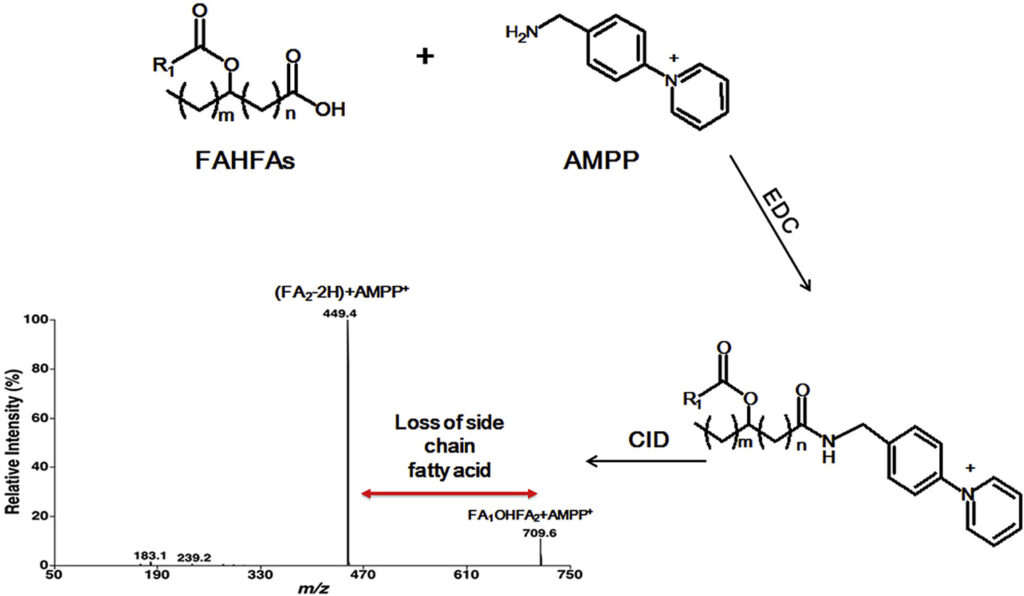
Branched fatty acid esters of hydroxy fatty acids (FAHFAs) are an important family of endogenous lipids, possessing antidiabetic and anti-inflammatory functions. Therefore, analysis of FAHFAs in biological samples obtained under healthy and disease states can uncover underlying mechanisms of various relevant disorders (e.g., diabetes and autoimmune diseases). Up to now, due to their extremely low abundance, the determination of the changed levels of these species is still a huge challenge, even though great efforts have been made by utilizing liquid chromatography-tandem mass spectrometry with or without derivatization. Herein, we described a novel method for analysis of FAHFAs present in lipid extracts of biological examples after solid-phase extraction and chemical derivatization with one authentic FAHFA specie as an internal standard based on the principles of multi-dimensional mass spectrometry-based shotgun lipidomics. The approach possessed marked sensitivity, high specificity, and broad linear dynamic range of over 3 orders without obvious matrix effects. Moreover, after chemical derivatization, the molecular masses of FAHFAs shift from an overlapped region with ceramide species to a new region without overlaps, removing these contaminating signals from ceramides, and thereby reducing the false results of FAHFAs. Finally, this novel method was successfully applied for determining FAHFAs levels in varieties of representative biological samples, including plasma from lean and overweight/obese individuals of normoglycemia, and tissue samples (such as liver and white adipose tissue from diabetic (db/db) mice). We revealed significant alterations of FAHFAs in samples under patho(physio)logical conditions compared to their respective controls. Taken together, the developed method could greatly contribute to studying altered FAHFA levels under a variety of biological/biomedical conditions, and facilitate the understanding of these lipid species in the patho(physio)logical process.
Graphical abstract
Advion Interchim Scientific® Triversa NanoMate® (Advion, Ithaca, NY) was utilized.
Abstract
Introduction
Agarwood is a highly valuable fragrant resinous wood which is widely used as traditional Chinese medicines, perfumes, incense and decorations. Due to its high economic value and excessive demand, this leads to a rising price and proliferation of fake commodities. Thus, strict authenticity identification and quality evaluation of agarwood are of great significance.
Objective
To establish a simple, rapid and non-destructive technique for identifying the authenticity of agarwood.
Methods
Liquid extraction surface analysis mass spectrometry (LESA-MS) was firstly proposed to identify the authenticity of 62 agarwood samples without sample preparation. In addition, multivariate statistical models and thin-layer chromatography (TLC) method were used to analyse and verify the results of LESA-MS.
Results
Representative compounds of agarwood were detected by LESA-MS. A characteristic 2-(2-phenylethyl)chromone compound (m/z 319.1) was treated as a key chemical marker to identify agarwood and its counterfeits rapidly. Several other chromones ions were identified and used as additional evidence for authentic samples. A total of 62 samples were visually discriminated as two groups by principal component analysis (PCA) and orthogonal projection to latent structures discriminant analysis (OPLS-DA), and the specific characteristic marker was highlighted. Moreover, the qualitative results of the conventional TLC method were in agreement with the LESA-MS approach.
Conclusion
The proposed LESA-MS method was successfully applied in the direct qualitative analysis of agarwood from different sources. This study indicated great feasibility and practicality of LESA-MS in the rapid identification of agarwood, and provided a non-destructive and meaningful preliminary screening tool for the agarwood industry.
Advion Interchim Scientific® Triversa NanoMate® (Advion, Ithaca, NY) was utilized.
Shaun N. Robertson, Fadi Soukarieh, Thomas M. White, Miguel Camara, Manuel Romero*, and Rian L. Griffiths*
Abstract
Previously, metabolites diffused or secreted from microbial samples have been analyzed via liquid chromatography–mass spectrometry (LC–MS) approaches following lengthy extraction protocols. Here, we present a model system for growing biofilms on discs before utilizing rapid and direct surface sampling MS, namely, liquid extraction surface analysis, to study the microbial exometabolome. One of the benefits of this approach is its surface-specific nature, enabling mimicking biofilm formation in a way that the study of planktonic liquid cultures cannot imitate. Even though Pseudomonas aeruginosa (P. aeruginosa), Staphylococcus aureus (S. aureus), and Candida albicans (C. albicans) have been studied previously in isolation, very few studies consider the complexity of the interplay between these pathogens, which are commonly combined causative agents of infection. Our model system provides a route to investigate changes in the exometabolome, such as metabolites that become circulatory in the presence of multiple pathogens. Our results agree with previous reports showing that 2-alkyl-4(1H)-quinolone signal molecules produced by P. aeruginosa are important markers of infection and suggest that methods for monitoring levels of 2-heptyl-4-hydroxyquinoline and 2,4-dihydroxyquinoline, as well as pyocyanin, could be beneficial in the determination of causative agents in interkingdom infection including P. aeruginosa. Furthermore, studying changes in exometabolome metabolites between pqs quorum sensing antagonists in treated and nontreated samples suggests suppression of phenazine production by P. aeruginosa. Hence, our model provides a rapid analytical approach to gaining a mechanistic understanding of bacterial signaling.
Advion Interchim Scientific® TriVersa® NanoMate® (Advion Interchim Scientific®, Ithaca, NY) was utilized for the LESA sampling.

Q: What is the focus of your lab’s research?
A: Our lab specializes in diagnostics and pathophysiology of rare genetic (metabolic) disease. Specifically, our work focuses on the development and introduction of untargeted metabolomics in diagnostics and on integrating genomics and metabolomics data to improve patient care. Together with our (inter)national collaborators we helped discover multiple novel genetic diseases in the past few years. The TriVersa® NanoMate® is used to infuse metabolite extracts of patient or research samples onto a high resolution mass spectrometer.
Q: Why did you incorporate the TriVersa® NanoMate® into your
laboratory?
A: The TriVersa® NanoMate® provides an easy to use and efficient method for sample infusion. Furthermore, and essential for our diagnostic practice, it virtually eliminates the chances of carryover.
Q: Who would you recommend to purchase the TriVersa® NanoMate®?
A: I recommend the TriVersa® NanoMate® to laboratories applying direct-infusion mass spectrometry for metabolomics (targeted and untargeted) analysis.
Q: Do you have any publications or presentations using the TriVersa® NanoMate®?
A: We performed untargeted metabolomics on dried blood spots of patients with 46 different genetic metabolic defects, and we simulated their whole-exome sequencing results in silico. We showed that for accurate prioritization of disease causing genes, it is essential to take into account not only the primary reaction of the affected protein but a larger network of potentially affected metabolites, multiple steps away from the primary reaction.
Cross-Omics: Integrating Genomics with Metabolomics in Clinical Diagnostics.
Kerkhofs MHPM, Haijes HA, Willemsen AM, van Gassen KLI, van der Ham M,
Gerrits J, de Sain-van der Velden MGM, Prinsen HCMT, van Deutekom HWM, van Hasselt PM, Verhoeven-Duif NM, Jans JJM. Metabolites. 2020 May 18;10(5):206.doi: 10.3390/metabo10050206.
Authors: Regensburg University Hospital, European Molecular Biology Laboratory, Germany; University of Southern Denmark, Denmark
Abstract
Lipidomics data require consideration of ions with near-identical masses, which comprises among others the Type-II isotopic overlap. This overlap occurs in series of lipid species differing only by number of double bonds (DBs) mainly because of the natural abundance of 13C-atoms. High-resolution mass spectrometry, such as Fourier-transform mass spectrometry (FTMS), is capable of resolving Type-II overlap depending on mass resolving power. In this work, we evaluated FTMS quantification accuracy of lipid species affected by Type-II overlap. Spike experiments with lipid species pairs of various lipid classes were analyzed by flow injection analysis-FTMS. Accuracy of quantification was evaluated without and with Type-II correction (using relative isotope abundance) as well as utilizing the first isotopic peak (M+1). Isobaric peaks, which were sufficiently resolved, were most accurate without Type-II correction. In cases of partially resolved peaks, we observed peak interference causing distortions in mass and intensity, which is a well-described phenomenon in FTMS. Concentrations of respective species were more accurate when calculated from M+1. Moreover, some minor species, affected by considerable Type-II overlap, could only be quantified by M+1. Unexpectedly, even completely unresolved peaks were substantially overcorrected by Type-II correction because of peak interference. The described method was validated including intraday and interday precisions for human serum and fibroblast samples. Taken together, our results show that accurate quantification of lipid species by FTMS requires resolution-depended data analysis.
Samples were infused using the Advion Interchim Scientific® TriVersa NanoMate®.
Authors: University of Graz, BioTechMed-Graz, Austria; Max Planck Institute of Molecular Cell Biology and Genetics, Germany
Abstract
Triacylglycerol (TG) and steryl ester (SE) lipid storage is a universal strategy to maintain organismal energy and membrane homeostasis. Cycles of building and mobilizing storage fat are fundamental in (re)distributing lipid substrates between tissues or to progress ontogenetic transitions. In this study, we show that Hormone-sensitive lipase (Hsl) specifically controls SE mobilization to initiate intergenerational sterol transfer in Drosophila melanogaster. Tissue-autonomous Hsl functions in the maternal fat body and germline coordinately prevent adult SE overstorage and maximize sterol allocation to embryos. While Hsl-deficiency is largely dispensable for normal development on sterol-rich diets, animals depend on adipocyte Hsl for optimal fecundity when dietary sterol becomes limiting. Notably, accumulation of SE but not of TG is a characteristic of Hsl-deficient cells across phyla including murine white adipocytes. In summary, we identified Hsl as an ancestral regulator of SE degradation, which improves intergenerational sterol transfer and reproductive success in flies.
Nano-ESI analysis was performed by chip-based infusion using the Advion Interchim Scientific® TriVersa NanoMate®.
Authors: University of Birmingham, United Kingdom
Abstract
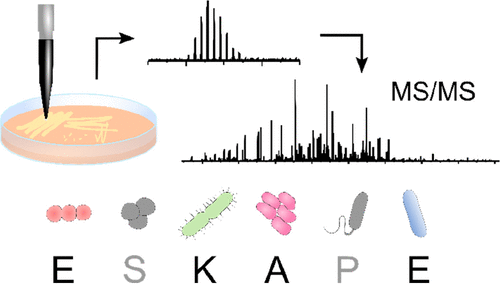
The ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae) represent clinically important bacterial species that are responsible for most hospital-acquired drug-resistant infections; hence, the need for rapid identification is of high importance. Previous work has demonstrated the suitability of liquid extraction surface analysis mass spectrometry (LESA-MS) for the direct analysis of colonies of two of the ESKAPE pathogens (Staphylococcus aureus and Pseudomonas aeruginosa) growing on agar. Here, we apply LESA-MS to the remaining four ESKAPE species (E. faecium E745, K. pneumoniae KP257, A. baumannii AYE, and E. cloacae S11) as well as E. faecalis V583 (a close relative of E. faecium) and a clinical isolate of A. baumannii AC02 using an optimized solvent sampling system. In each case, top-down LESA MS/MS was employed for protein identification. In total, 24 proteins were identified from 37 MS/MS spectra by searching against protein databases for the individual species. The MS/MS spectra for the identified proteins were subsequently searched against multiple databases from multiple species in an automated data analysis workflow with a view to determining the accuracy of identification of unknowns. Out of 24 proteins, 19 were correctly assigned at the protein and species level, corresponding to an identification success rate of 79%.
LESA-MS was performed using the Advion Interchim Scientific® TriVersa NanoMate®.
